Covering All Your Bases: Strategies to Expand Training Data for Specialized Genomics Problems
Authors:




This work was done during Ren Yi’s internship at Google.
We would like to thank Babak Alipanahi and Maria Nattestad for editorial suggestions and review.
This blog post summarizes work accepted to the Machine Learning 4 Health (ML4H) workshop at Neural Information Processing Systems (NeurIPS 2019). The full paper, titled Learning from Data-Rich Problems: A Case Study on Genetic Variant Calling, is available on arXiv and goes into greater technical detail.
Introduction
Training machine learning models involves tension between capturing true concepts that generalize to solve a problem and identifying the patterns in a training set which do not, referred to as overfitting. Large, comprehensive training datasets lead to better accuracy as they dilute spurious correlations and are more likely to contain multiple examples of difficult edge cases.
In some domains, there is a large body of training data for a given problem, while a slightly different problem has substantially fewer labeled datasets. In this case, we have a dilemma: how can we train a model that captures the specialized aspects of a data-limited problem and benefits from large amounts of related training data? For machine learning applications where gathering more data is infeasible, transfer learning can be used for learning general concepts from similar problems with abundant data. For example, image classification models are commonly pretrained using the large ImageNet dataset, prior to training on a smaller task-specific dataset.
Two such related problems in genomics are variant calling using whole genome sequencing (WGS) data and variant calling using whole exome sequencing (WES) data, which targets protein-coding regions that make up 1-2% of the genome. The high-quality Genome in a Bottle (GIAB) truth sets, which enable training of methods like Clairvoyante and DeepVariant, contain ~4,000,000 variant examples per individual in WGS but only ~200,000 for typical WES captures.
Our team has publicly released three DeepVariant models: Illumina WGS, PacBio WGS, and Illumina WES. Historically, WES models have been more difficult to train due to the reduced number of training examples and achieve lower accuracy than the WGS models. The lower accuracy is also attributed, at least in part, to the overall greater difficulty in analyzing exomes due to additional variability in coverage, capture efficiency, and the use of PCR, highlighting the need for a specialized model. Nonetheless, WGS data shares many broad similarities with WES data, and models may capture high-level concepts from WGS data that generalize to WES data. In this work, we use DeepVariant to explore three different training strategies that leverage WGS data for improving performance on WES data.
Methods
Below are the three methods we considered for this work. For each approach, HG001 samples are used for training and tuning. Tune sets used for picking all model checkpoints consist of WES data from chr1, which was excluded during training. Evaluation is performed on WES data from HG002. Train, tune, and evaluation labels come from GIAB truth sets.
1. Jointly train with both WGS and WES data, select a model using tune performance on WES

2. Warmstart from a pre-trained WGS model and train on WES data to adapt to it

We first train a model using WGS data, and then finetune using only WES data. This is similar to the approach used to train the production WES and PacBio models.
3. Add an additional vector within the model architecture to capture sequencing type

We train jointly with WGS and WES data, similar to the first method. However, we add a vector (SeqType) that indicates the type of each example as either WES or WGS. This feature is added near the end of the neural network. In this case, we provide only WES or WGS as inputs, but this approach could be extended to include other features, such as instrument type (NovaSeq vs. HiSeq) or sample preparation (PCR-Free vs PCR+).
Data
In this investigation, we use a subset of the datasets used for training the production DeepVariant models. We do this in order to iterate quickly on model training, enabling the project to conclude within the span of an internship. In addition, decreasing the size of the training data is useful to reduce the number of confounding factors, such as differences in sequencer or sample preparation method. Though these results are generally informative about training strategies, we have not yet incorporated them into the training of production releases of DeepVariant.
Our experimental dataset contains three Illumina HiSeq2500 PCR-Free WGS BAM files and 18 Illumina HiSeq4000 WES BAM files. In comparison, the production DeepVariant models are trained using ~8-9 fold additional data from HiSeqX, NovaSeq, and PCR+ samples, and different WES capture kits.
Number of examples in training DeepVariant models for this investigation:
| WGS | WES | |
|---|---|---|
| Train | 37,106,930 | 2,641,013 |
| Tune | 1,024,080 | 94,149 |
Number of examples in training production DeepVariant models:
| WGS | WES | |
|---|---|---|
| Train | 320,662,815 | 17,402,861 |
| Tune | 2,435,712 | 631,261 |
Results
Overall performance of each approach is shown, as measured by F1 on WES data. All reported numbers are from HG002, which was not seen during training. We include a baseline of training only with WES data (WES Only).
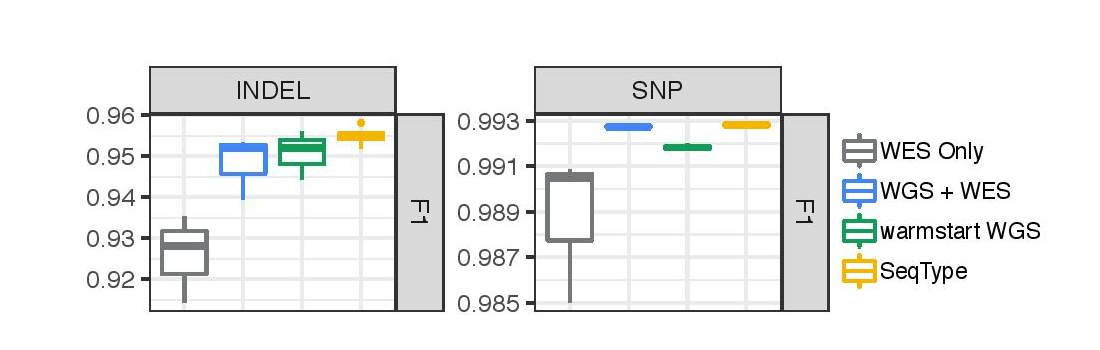
All of the training strategies outperform the WES Only baseline, especially for Indel performance. The SeqType approach performs the best overall, outperforming other methods on both SNP and Indel accuracy.
There is an interesting trade-off between the WGS + WES method versus the warmstart WGS method. Training with both WGS and WES results in better performance on SNPs, while warmstarting followed by training only on WES results in better performance on Indels. One possible reason for this may be that Indel calling differs more between WES and WGS data than SNP calling. When the final dataset used for training consists only of exomes, the model adapts more completely to the specialized components of the Indel problem at the cost of some of the general learnings of the larger SNP problem. The benefit of SeqType may come from allowing both data types to be exposed while giving the means to distinguish the specialized aspects of the exome problem.
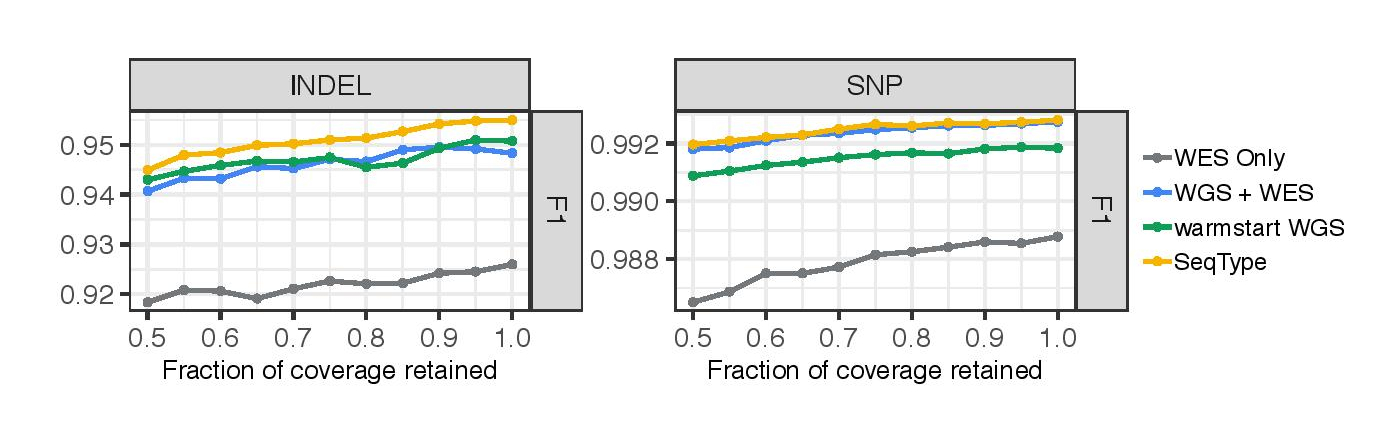
In addition to conducting the experiment at full coverage, we also assessed each approach on exomes with serially downsampled coverage. Overall, we observe the same trend between the models across a range of coverages (roughly ranging from 100x - 200x capture coverage).
Conclusion
We demonstrate three approaches that use WGS data to improve model performance for the specialized task of variant calling from WES data. Supervised pretraining and joint training strategies such as these can also be applied to other problems in genomics. For example, we may wish to pretrain models using Illumina data prior to fine tuning on long-read data from newer sequencing technologies, such as PacBio or Oxford Nanopore. In addition to supervised methods, recent work has shown how unsupervised methods using large protein databases can be used to improve performance on downstream tasks, for which there exists limited data.
While we currently train separate models for three different data types (WGS, WES, PacBio), the SeqType approach provides a framework through which a more general model could be developed. This approach could naturally be extended to other characteristics of the data, such as sequencer (HiSeq2500 vs. HiSeqX vs. NovaSeq) and sample preparation (PCR-Free vs. PCR+). Currently, we do not explicitly include these features as inputs, but doing so might allow for the development of a unified model that performs well across various different data types.
As a final reminder, this work was conducted by Ren Yi during her internship at Google. Ren is currently a PhD student at NYU and will present this work at the Machine Learning 4 Health (ML4H) workshop at Neural Information Processing Systems (NeurIPS 2019). If you would like to learn more about this work, please see the arXiv paper: Learning from Data-Rich Problems: A Case Study on Genetic Variant Calling.