This space contains public investigations and discussions from The Genomics team in Google Health. Our intended audience is the genomics community, and those within the machine learning community with a desire to learn about application within genomics.
-
Using AI to identify genetic variants in tumors with DeepSomatic → [External: Google Research Blog]
 DeepSomatic is an AI-powered tool that identifies cancer-related mutations in a tumor’s genetic sequence to help pinpoint what’s driving the cancer.
DeepSomatic is an AI-powered tool that identifies cancer-related mutations in a tumor’s genetic sequence to help pinpoint what’s driving the cancer. -
10 years of genomics research at Google → [External: The Keyword | Google]
 A look back at a decade of research in genomics at Google, from developing AI tools that accurately read the code of life, creating comprehensive reference genomes and working with partners on real-world challenges from biodiversity to healthcare.
A look back at a decade of research in genomics at Google, from developing AI tools that accurately read the code of life, creating comprehensive reference genomes and working with partners on real-world challenges from biodiversity to healthcare. -
Highly accurate genome polishing with DeepPolisher: Enhancing the foundation of genomic research → [External: Google Research Blog]
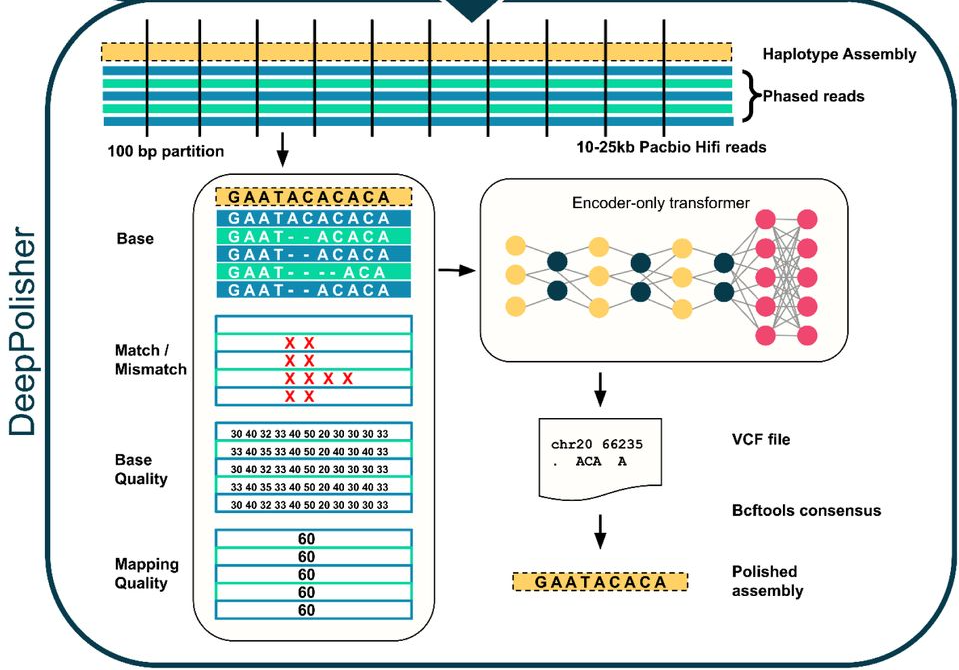 DeepPolisher, is a new deep learning tool that significantly improves the accuracy of genome assemblies by precisely correcting base-level errors, which recently played a key role in enhancing the Human Pangenome Reference.
DeepPolisher, is a new deep learning tool that significantly improves the accuracy of genome assemblies by precisely correcting base-level errors, which recently played a key role in enhancing the Human Pangenome Reference. -
Learning DeepVariant's Hidden Powers
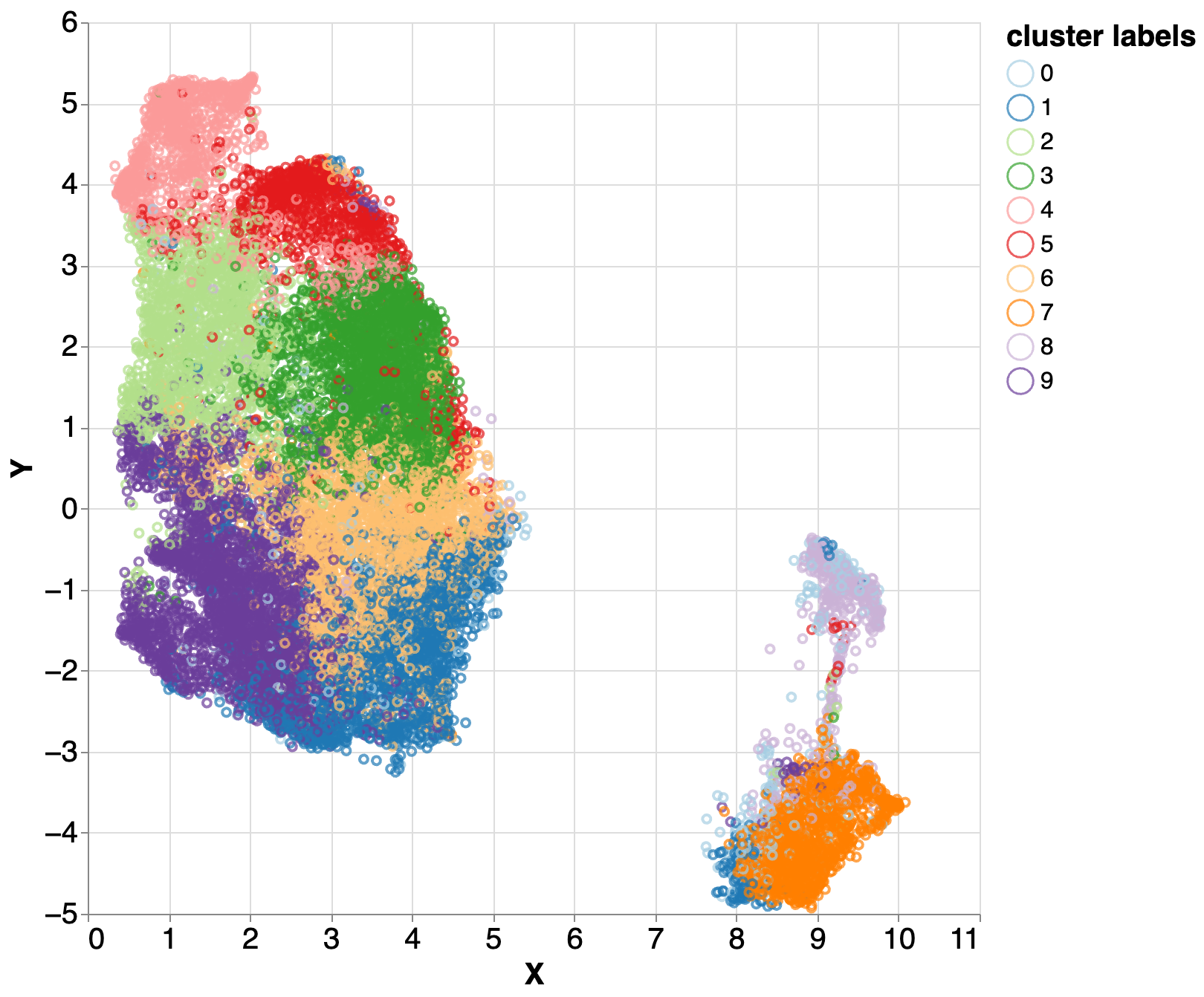 We examined the DeepVariant model to determine what insights it has developed, and we discovered that it utilizes insights generally applied in other tasks but on which it is not trained.
We examined the DeepVariant model to determine what insights it has developed, and we discovered that it utilizes insights generally applied in other tasks but on which it is not trained. -
Blindfolding DeepVariant: Surprising Insights from Hiding Information
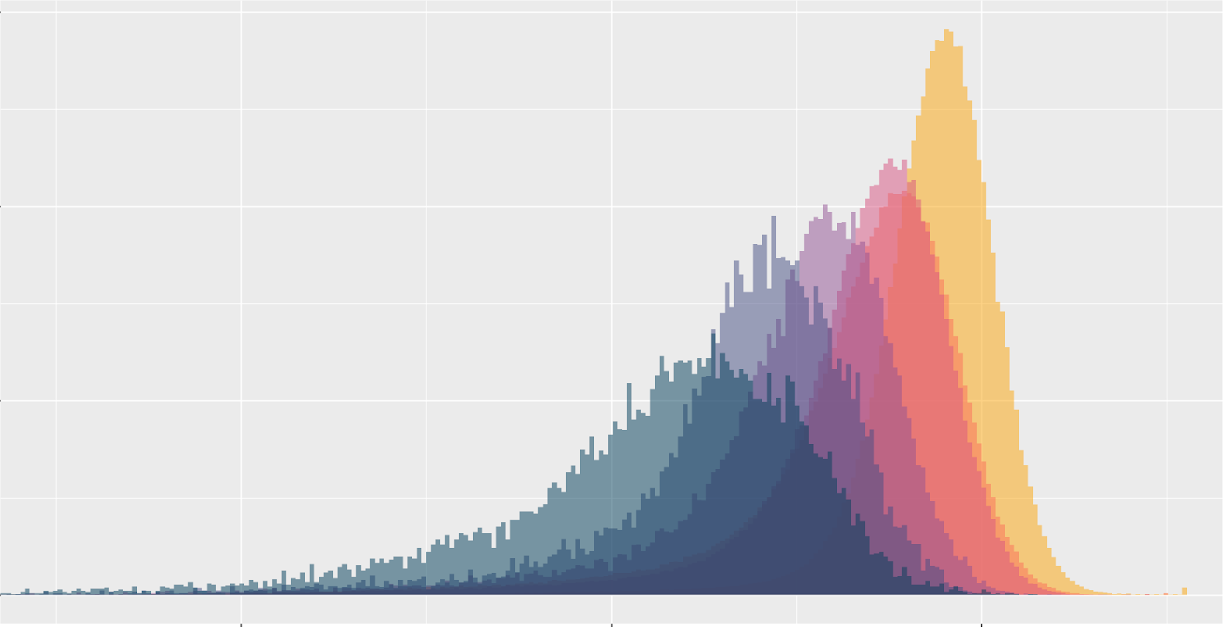 This post investigates the importance of various features of sequencing data to the ability to accurately call variants. Channel ablation experiments reveal how DeepVariant uses specific representational information to correctly call specific genotypes.
This post investigates the importance of various features of sequencing data to the ability to accurately call variants. Channel ablation experiments reveal how DeepVariant uses specific representational information to correctly call specific genotypes. -
Building better pangenomes to improve the equity of genomics → [External: Google Research Blog]
 In this post, we discuss the methods development that contributed to building and using Pangenome references in the paper “A draft human pangenome reference”. We outline the rationale for using pangenomes. We discuss how accuracy in the sequences which compose Pangenomes is essential, and how DeepConsensus can be used to improve sequence for pangenomes and DeepVariant was adapted to polish pangenome assemblies. We discuss methods that map sequences to pangenomes to achieve higher accuracy.
In this post, we discuss the methods development that contributed to building and using Pangenome references in the paper “A draft human pangenome reference”. We outline the rationale for using pangenomes. We discuss how accuracy in the sequences which compose Pangenomes is essential, and how DeepConsensus can be used to improve sequence for pangenomes and DeepVariant was adapted to polish pangenome assemblies. We discuss methods that map sequences to pangenomes to achieve higher accuracy. -
An ML-based approach to better characterize lung diseases → [External: Google Research Blog]
 In this post, we highlight a new machine-learning-based phenotyping approach published in Nature Genetics. COPD liability scores from our deep learning model improve genetic association discovery and risk prediction. We trained our model using high dimensional lung function data and noisy medical record labels obtained from self-reporting and hospital diagnostic codes, and demonstrated that our method generalizes to diseases that lack expert-defined annotations.
In this post, we highlight a new machine-learning-based phenotyping approach published in Nature Genetics. COPD liability scores from our deep learning model improve genetic association discovery and risk prediction. We trained our model using high dimensional lung function data and noisy medical record labels obtained from self-reporting and hospital diagnostic codes, and demonstrated that our method generalizes to diseases that lack expert-defined annotations. -
Developing an aging clock using deep learning on retinal images → [External: Google Research Blog]
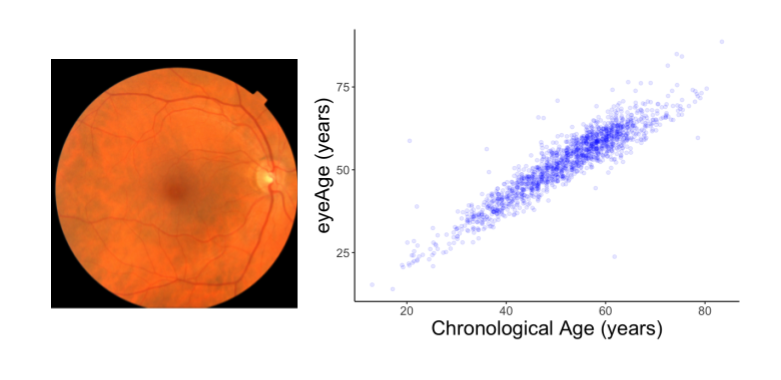 In this post we develop a deep learning model to predict biological age from retinal images. We show that a gene highly associated with accelerated biological aging contributes to age-related vision loss in flies, providing a proof-of-concept for using machine learning to discover genetic targets for age-related diseases.
In this post we develop a deep learning model to predict biological age from retinal images. We show that a gene highly associated with accelerated biological aging contributes to age-related vision loss in flies, providing a proof-of-concept for using machine learning to discover genetic targets for age-related diseases. -
Variational inference for polygenic risk scores
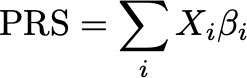 We introduce a new way to build polygenic risk scores from GWAS summary statistics using black box variational inference.
We introduce a new way to build polygenic risk scores from GWAS summary statistics using black box variational inference. -
Adding Custom Channels to DeepVariant
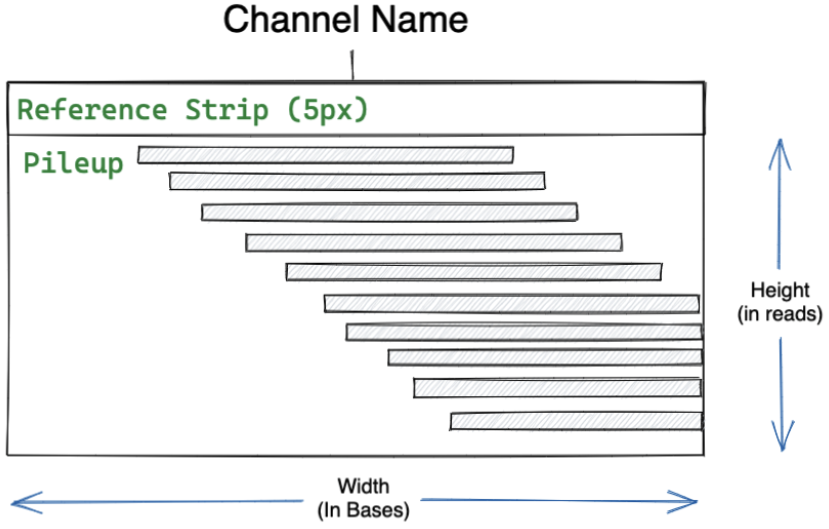 This tutorial will show you how to add custom channels to DeepVariant, train new models, and use them to perform variant calling. Custom channels can be used to tailor DeepVariant to a specific sequencing platform or applications.
This tutorial will show you how to add custom channels to DeepVariant, train new models, and use them to perform variant calling. Custom channels can be used to tailor DeepVariant to a specific sequencing platform or applications. -
DeepNull: an open-source method to improve the discovery power of genetic association studies → [External: Google Open Source Blog]
 In this post, we describe a new method, DeepNull, which models the complex relationship between covariate effects on phenotypes to improve Genome-wide association studies (GWAS) results. We discuss why correcting for the relationships is important, and how DeepNull is able to better do so.
In this post, we describe a new method, DeepNull, which models the complex relationship between covariate effects on phenotypes to improve Genome-wide association studies (GWAS) results. We discuss why correcting for the relationships is important, and how DeepNull is able to better do so. -
Advancing genomics to better understand and treat disease → [External: The Keyword | Google]
 At Google Health, we’re applying our technology and expertise to the field of genomics. Here are recent research and industry developments we’ve made to help quickly identify genetic disease and foster the equity of genomic tests across ancestries. This includes an exciting new partnership with Pacific Biosciences to further advance genomic technologies in research and the clinic.
At Google Health, we’re applying our technology and expertise to the field of genomics. Here are recent research and industry developments we’ve made to help quickly identify genetic disease and foster the equity of genomic tests across ancestries. This includes an exciting new partnership with Pacific Biosciences to further advance genomic technologies in research and the clinic. -
Improving Genomic Discovery with Machine Learning → [External: Google AI Blog]
 In this post, we demonstrate how using ML models to classify medical imaging data can be used to improve GWAS. We describe how models can be trained for phenotypes to generate trait predictions, how these predictions are used to identify novel genetic associations, and that the novel associations discovered improve PRS accuracy.
In this post, we demonstrate how using ML models to classify medical imaging data can be used to improve GWAS. We describe how models can be trained for phenotypes to generate trait predictions, how these predictions are used to identify novel genetic associations, and that the novel associations discovered improve PRS accuracy. -
DeepVariant over the years
 In this post, we summarize the improvements in accuracy and runtime over the years and highlight a few categories of changes that have led to these improvements.
In this post, we summarize the improvements in accuracy and runtime over the years and highlight a few categories of changes that have led to these improvements. -
Analyzing genomic data in families with deep learning → [External: Google Open Source Blog]
 This post dives into DeepTrio, which can jointly analyze a mother-father-child trio of samples. We discuss how we represent the trio data and how DeepTrio is trained. We give accuracy benchmarks for DeepTrio, showing that it has higher accuracy than single sample calling, especially at low sequence depths.
This post dives into DeepTrio, which can jointly analyze a mother-father-child trio of samples. We discuss how we represent the trio data and how DeepTrio is trained. We give accuracy benchmarks for DeepTrio, showing that it has higher accuracy than single sample calling, especially at low sequence depths. -
Improving Variant Calling using Haplotype Information
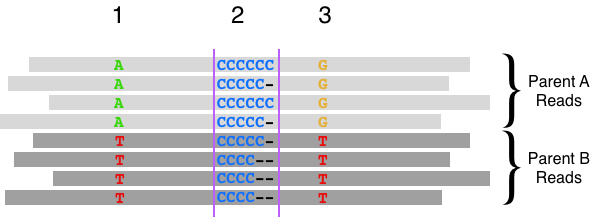 We discuss a new channel in DeepVariant which encodes haplotype information in long-read data, and was released with DeepVariant v1.1. We review how haplotypes relate to variant calling, show examples improved by the channel, and quantify the accuracy improvement with PacBio HiFi reads.
We discuss a new channel in DeepVariant which encodes haplotype information in long-read data, and was released with DeepVariant v1.1. We review how haplotypes relate to variant calling, show examples improved by the channel, and quantify the accuracy improvement with PacBio HiFi reads. -
Improving the Accuracy of Genomic Analysis with DeepVariant 1.0 → [External: Google AI Blog]
 This post covers the release of DeepVariant v1.0, which incorporates a large number of improvements for all sequencing types. DeepVariant v1.0 is an improved version of our submission to the PrecisionFDA v2 Truth Challenge, which achieved Best Overall accuracy for 3 of 4 instrument categories.
This post covers the release of DeepVariant v1.0, which incorporates a large number of improvements for all sequencing types. DeepVariant v1.0 is an improved version of our submission to the PrecisionFDA v2 Truth Challenge, which achieved Best Overall accuracy for 3 of 4 instrument categories. -
Looking Through DeepVariant's Eyes
 DeepVariant turns variant-calling into an image classification task. Here we explore what these pileup images look like and try to do the same classification task ourselves. We show easy and difficult examples, including multiallelics. By the end, we have a better intuition for how DeepVariant works.
DeepVariant turns variant-calling into an image classification task. Here we explore what these pileup images look like and try to do the same classification task ourselves. We show easy and difficult examples, including multiallelics. By the end, we have a better intuition for how DeepVariant works. -
Covering All Your Bases: Strategies to Expand Training Data for Specialized Genomics Problems
 We explore three different training strategies to leverage whole-genome sequencing data to improve model performance for the specialized task of variant calling from whole-exome sequencing data: 1) jointly training with both WGS and WES data, 2) warmstarting from a pre-trained WGS model, and 3) including sequencing type as an input to the model.
We explore three different training strategies to leverage whole-genome sequencing data to improve model performance for the specialized task of variant calling from whole-exome sequencing data: 1) jointly training with both WGS and WES data, 2) warmstarting from a pre-trained WGS model, and 3) including sequencing type as an input to the model. -
Twenty is the new Thirty - Comparing Current and Historical WGS Accuracy Across Coverage
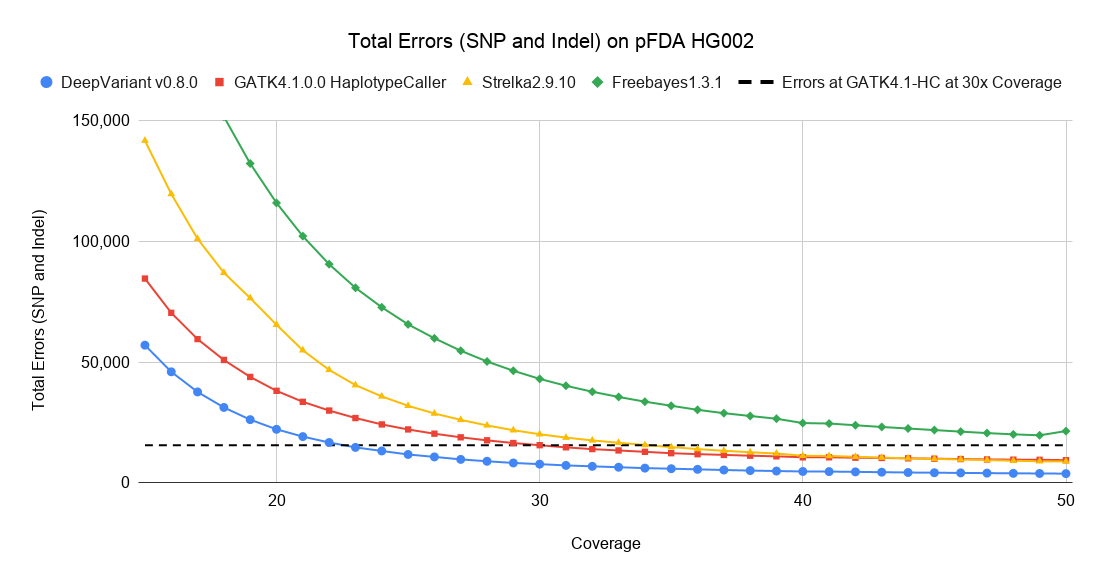 In this blog, we discuss how sequencing coverage involves trade-offs between cost and accuracy. We explore how computational methods that improve accuracy can also be understood as reducing cost. We compare current methods to historical accuracies. Finally, we explore the types of errors present at low and high coverages.
In this blog, we discuss how sequencing coverage involves trade-offs between cost and accuracy. We explore how computational methods that improve accuracy can also be understood as reducing cost. We compare current methods to historical accuracies. Finally, we explore the types of errors present at low and high coverages. -
The Power of Building on an Accelerating Platform: How DeepVariant Uses Intel’s AVX-512 Optimizations
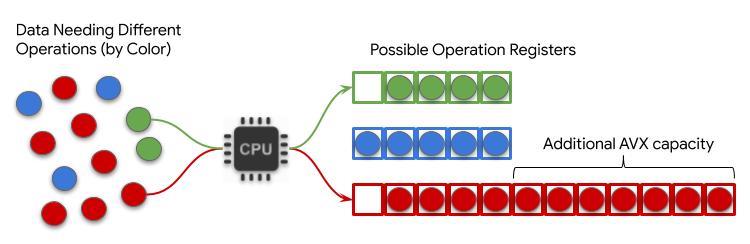 The v0.7 release of DeepVariant featured a three-fold improvement in end-to-end speed and a corresponding decrease in cost relative to the previous version (v0.6). Much of this speed improvement comes by enabling DeepVariant to take advantage of new Intel® Advanced Vector eXtensions (AVX-512) instruction set.
The v0.7 release of DeepVariant featured a three-fold improvement in end-to-end speed and a corresponding decrease in cost relative to the previous version (v0.6). Much of this speed improvement comes by enabling DeepVariant to take advantage of new Intel® Advanced Vector eXtensions (AVX-512) instruction set. -
Analyzing 3024 rice genomes characterized by DeepVariant → [External: Google Cloud Blog]
 This post explores how to identify and analyze different rice genome mutations with DeepVariant. To do this, we performed a re-analysis of the Rice 3K dataset and have made the data publicly available as part of the Google Cloud Public Dataset Program.
This post explores how to identify and analyze different rice genome mutations with DeepVariant. To do this, we performed a re-analysis of the Rice 3K dataset and have made the data publicly available as part of the Google Cloud Public Dataset Program. -
Using Nucleus and TensorFlow for DNA Sequencing Error Correction
 An example of how Nucleus, a library for reading, writing, and processing genomics data, can be used alongside TensorFlow for machine learning. We discuss two approaches for the problem of DNA sequencing error correction and implement the second approach in this Colab tutorial.
An example of how Nucleus, a library for reading, writing, and processing genomics data, can be used alongside TensorFlow for machine learning. We discuss two approaches for the problem of DNA sequencing error correction and implement the second approach in this Colab tutorial. -
Highly Accurate SNP and Indel Calling on PacBio CCS with DeepVariant
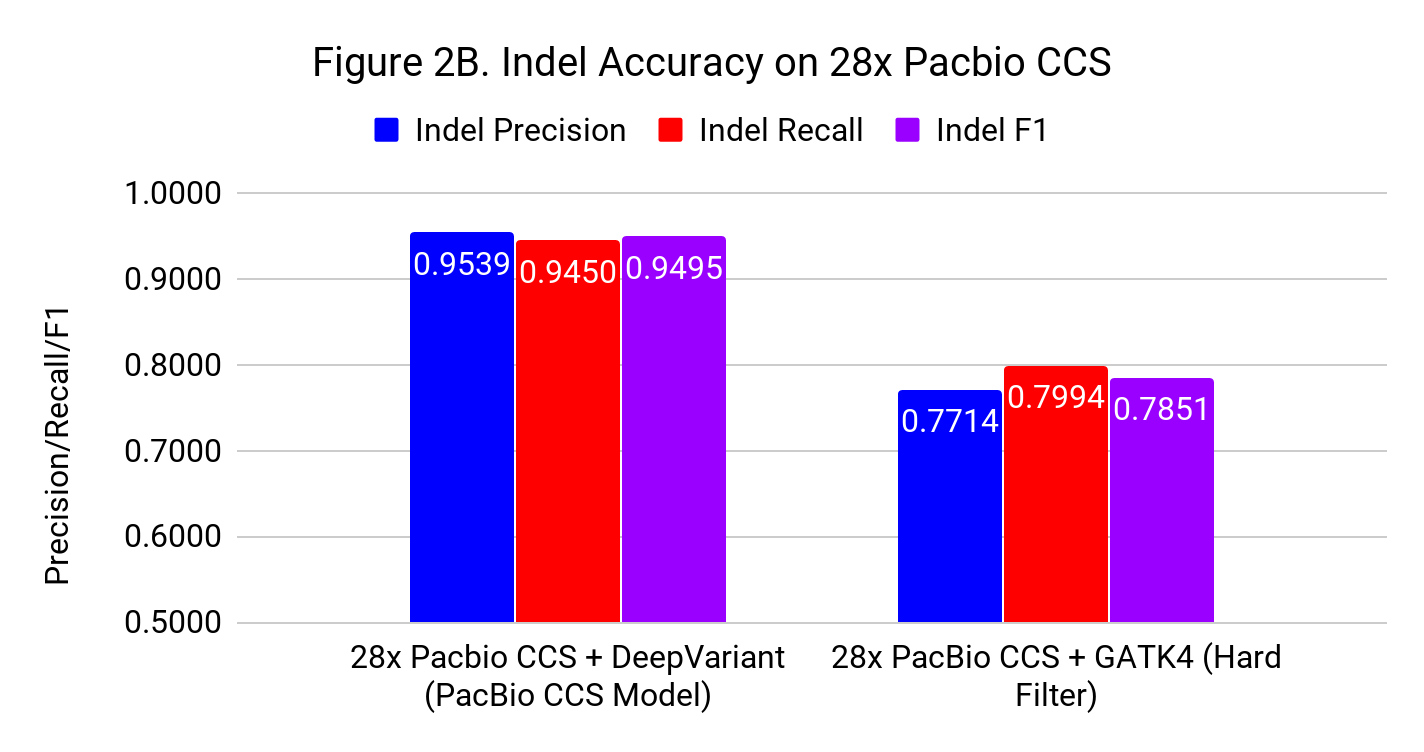 We discuss the newly published use of PacBio Circular Consensus Sequencing (CCS) at human genome scale. DeepVariant trained for this data type achieves similar accuracy to available Illumina genomes, and is the only method to achieve competitive accuracy in Indel calling. Early access to this model is available now by request, and we expect general availability in our next DeepVariant release (v0.8).
We discuss the newly published use of PacBio Circular Consensus Sequencing (CCS) at human genome scale. DeepVariant trained for this data type achieves similar accuracy to available Illumina genomes, and is the only method to achieve competitive accuracy in Indel calling. Early access to this model is available now by request, and we expect general availability in our next DeepVariant release (v0.8). -
Improved non-human variant calling using species-specific DeepVariant models
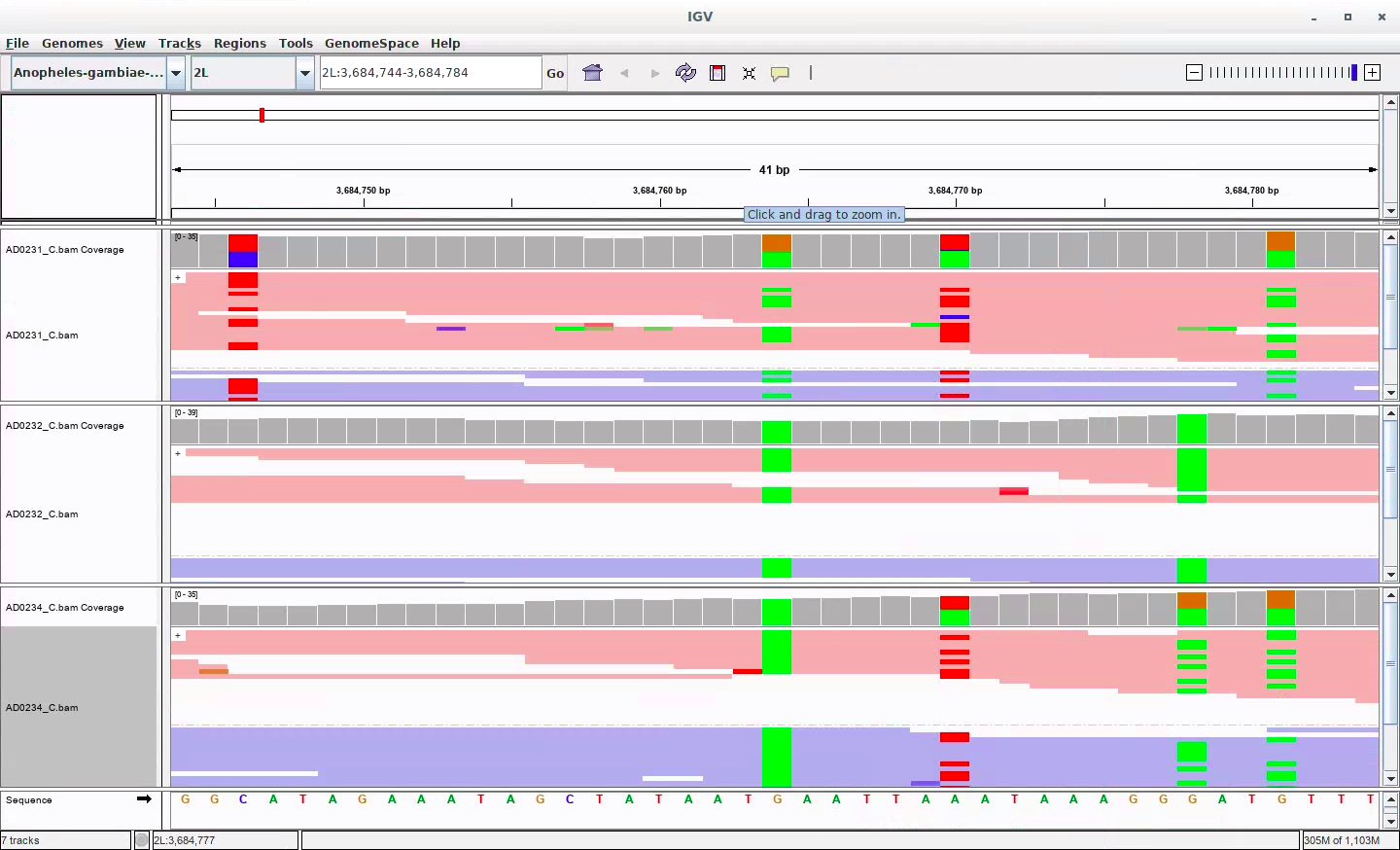 We investigate variant calling across a pedigree of mosquito genomes. Using rates of Mendelian violation, we assess pipelines developed to call variation in humans when applied to mosquito samples.
We investigate variant calling across a pedigree of mosquito genomes. Using rates of Mendelian violation, we assess pipelines developed to call variation in humans when applied to mosquito samples. -
DeepVariant Accuracy Improvements for Genetic Datatypes → [External: Google AI Blog]
 This post covers the release of DeepVariant v0.6, which includes some major accuracy improvements. We describe how we train DeepVariant, and how we were able to improve DeepVariant's accuracy for two common sequencing scenarios, whole exome sequencing and polymerase chain reaction sequencing, simply by adding representative data into DeepVariant's training process.
This post covers the release of DeepVariant v0.6, which includes some major accuracy improvements. We describe how we train DeepVariant, and how we were able to improve DeepVariant's accuracy for two common sequencing scenarios, whole exome sequencing and polymerase chain reaction sequencing, simply by adding representative data into DeepVariant's training process. -
DeepVariant: Highly Accurate Genomes With Deep Neural Networks → [External: Google AI Blog]
 We announce the open source release of DeepVariant, a deep learning technology to reconstruct the true genome sequence from HTS sequencer data with significantly greater accuracy than previous classical methods.
We announce the open source release of DeepVariant, a deep learning technology to reconstruct the true genome sequence from HTS sequencer data with significantly greater accuracy than previous classical methods.