Twenty is the new Thirty - Comparing Current and Historical WGS Accuracy Across Coverage
Authors:


Editorial suggestions and review: Maria Nattestad, Gunjan Baid, Taedong Yun
In this blog, we discuss how sequencing coverage involves trade-offs between cost and accuracy. We explore how computational methods that improve accuracy can also be understood as reducing cost. We compare current methods to historical accuracies. Finally, we explore the types of errors present at low and high coverages.
Sequencing coverage refers to how many times an instrument has sampled the content of a genome. If a chromosome could be chosen at will and sequenced without error, a diploid organism would require a coverage of 2 to perfectly analyze. However, sequencing is prone to random and systematic errors from both instruments and algorithms, differences in sampling efficiency for certain genome contexts, and random sampling of parental chromosomes.
As a result, higher coverages allow more confident analysis of a genome. Generating coverage uses reagents and instrument time, creating trade-offs between cost and accuracy. Exome and panel sequencing also manage cost by limiting coverage to specific parts of the genome.
Early in the history of sequencing projects at scale, the field converged around the concept that 30-fold coverage represents the ideal trade-off of accuracy and cost for what is considered a “high quality” genome. This concept has become deeply ingrained in the community mindset, even as the sequencing and analysis fields have evolved rapidly.
We have previously discussed DeepVariant’s accuracy in the context of reducing errors and performing robustly across data types. In this blog, we reframe the question of accuracy through the perspective of cost. Specifically, given DeepVariant’s accuracy, how much cost could be saved in coverage while still achieving community expectations on accuracy.
In this analysis, we use the 50-fold PCR-Free HG002 sample used in the PrecisionFDA Truth Challenge. DeepVariant is never trained on this sample. We randomly downsample this in increments of 1-fold coverage to create 15-fold to 50-fold coverage titrations. This assessment uses the Genome in a Bottle 3.3.2 Truth Set from NIST.
Assessing total errors
First, we look at the total number of errors of all types as a function of coverage. This covers both SNP and Indel errors and combines false positives, false negatives, and genotype errors (which are counted only once). To frame the number of errors in context, we also plot the number of errors for several submissions to the 2016 PrecisionFDA Truth Challenge, which was conducted on the same data as the 50x coverage point.
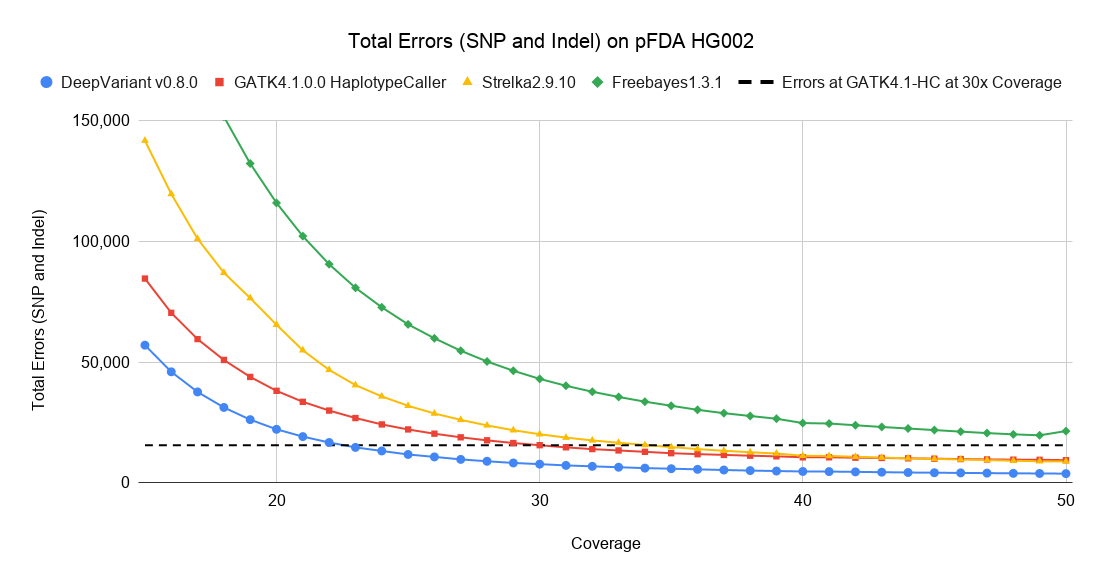
In this sample, DeepVariant at around 22x-23x coverage has the same number of errors as GATK4-HC at 30x coverage. If we consider a 30x genome to cost $1,000 and cost linearly scales with coverage, this corresponds to a potential saving of $250 to achieve the same number of errors.
The number of errors continues to decrease as coverage increases past 30x. The effective coverage gap between DeepVariant and GATK4-HC grows, but both methods start to reach an accuracy plateau. DeepVariant at 27x coverage has about the same number of errors as GATK4-HC at 50x coverage (a savings of ~$766 for the 50x data point).
Current accuracies compared to historical performance
The pipelines in the above investigation represent current performance in 2019. How does this compare to the historical performance of pipelines? To assess this, we used several submissions to the 2016 Truth Challenge: a GATK Best Practices submission, GATK with VQSR, Deepak Grover’s submission which used GATK plus custom analysis and was awarded best Indel accuracy, and the proof-of-concept version DeepVariant submission.
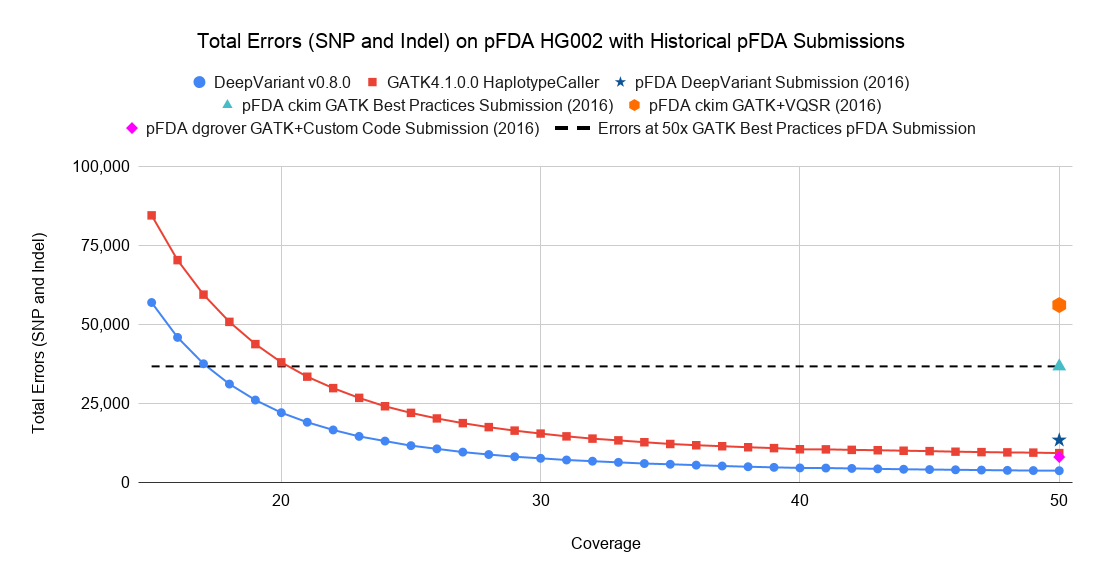
Relative to the GATK Best Practices and Best Practices with VQSR submissions, the recent runs of both GATK4 and DeepVariant are much higher in accuracy. Here the current DeepVariant is able to match the 50x GATK Best Practices submission at a coverage of around 17x.
There are two possible explanations. Accuracies as a whole may have improved substantially since 2016 (DeepVariant’s accuracy has continued to improve through the open-source release and subsequent versions). Alternatively, many of the Best Practices submissions may not have been executed in an ideal manner (we note that other pFDA GATK submissions have a similar error number to the one chosen in this chart). Because GATK3 requires a license for use by commercial entities, it is difficult for us to check these two possibilities ourselves.
Whichever possibility is correct, this observation is motivation to assess the accuracy of your pipeline with Genome in a Bottle. If your analysis pipeline has not been updated in several years, it is likely worth considering what you could achieve in cost or accuracy with newer methods (and we hope that when you do so you will consider DeepVariant for your use cases).
Assessing error types across coverage ranges
Next we investigate how variant callers balance between false positives and negatives as coverage drops. The next two charts break down false positives (reference sites called as a variant of any genotype), false negatives (variant sites of any genotype called as reference), and genotype errors (heterozygous variants misclassified as homozygous and vice versa) for SNPs and Indels.
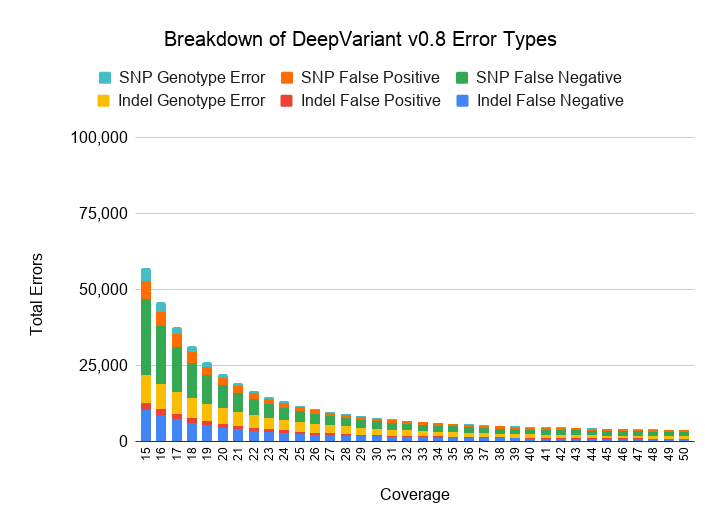
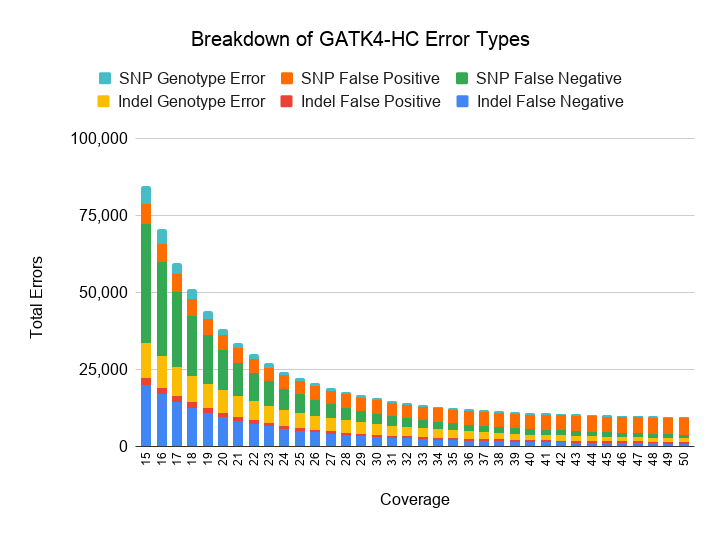
We also plot the relative proportion of each error class for DeepVariant v0.8 and GATK4-HC.
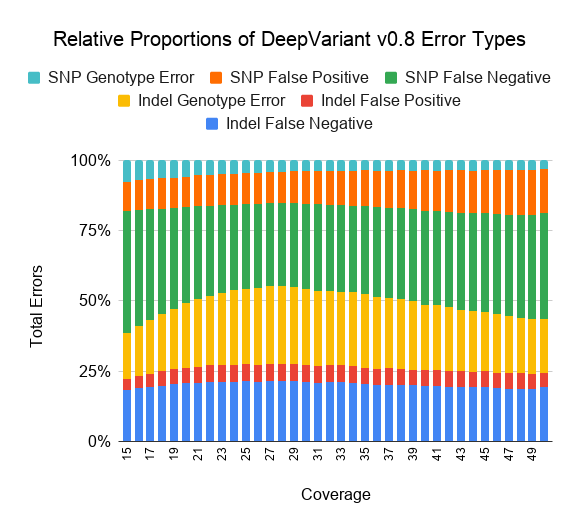
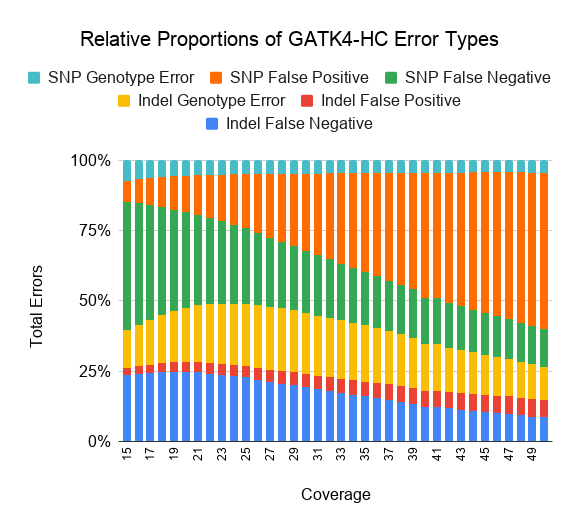
As coverages decreases from 30x, both GATK4-HC and DeepVariant v0.8 behave in qualitatively similar ways. False negatives increase with lower coverage, especially for SNPs at less than 20x coverage.
At high coverages, GATK4-HC and DeepVariant diverge in error profile. DeepVariant maintains a similar proportion of false positives and false negatives at higher coverages, while for GATK, higher coverage mostly does not decrease its false positive rate and instead reduces its false negatives.
Genomic regions contributing to differences in accuracy
To understand the differences across coverages, we looked into stratifications of accuracy across genomic regions from the extended evaluation of hap.py. There are a very large number of potential comparisons, so we will highlight only a few regions.
The chart below plots how much WGS coverage is required for the error number of GATK4-HC and DeepVariant v0.8.0 to be roughly equal determined with the 35 WGS samples titrated from 15x to 50x.
For example, we highlight with a blue star the data point indicating that 22x coverage of DeepVariant has a similar number of errors to 29x of GATK4-HC when considering all truth set sites.
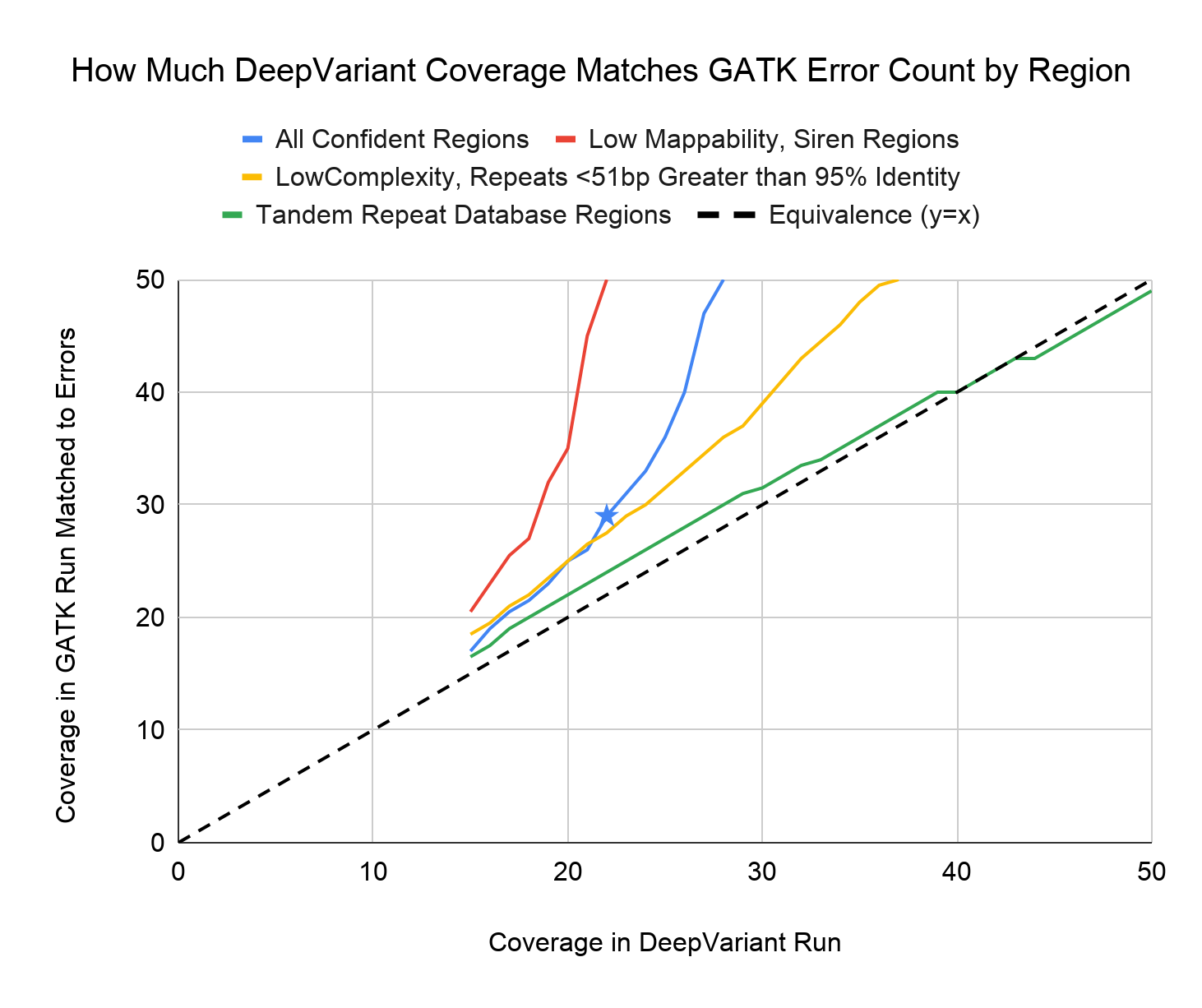
As the chart indicates, some regions are disproportionately more accurate for DeepVariant, even when compared with its overall higher accuracy. We observe that the stratification region for low mappability, siren regions contribute to improved accuracy at lower coverages.
At the same time, we are also able to identify some regions where the number of errors between DeepVariant and GATK are more similar. Areas near tandem repeats stand out in this category. We have investigated what may be occurring here and our early conclusion is that DeepVariant’s human-coded sensitive caller which identifies candidate positions sometimes does not identify these positions in short read data. Based on that observation, we are looking into ways that improve candidate generation near tandem repeats to improve DeepVariant’s accuracy in these regions.
Conclusion
Next Generation Sequencing is rapidly growing, but still not universal in research and diagnostics. For technologies in this state, the decision of whether to use it must weigh benefits versus costs. Our goal in developing DeepVariant is to support applications at each point of the constraint curve of cost, accuracy, and input types. We train across a coverage range of 20x-60x, in addition to training for performance on PCR-positive sequencing preparations, exomes, and for PacBio HiFi data.
As sequencing and the analytical tools for it evolve, we hope that the community will constantly re-evaluate the benefits and constraints. Perhaps sequencing at lower coverage will allow broadening studies or access. Perhaps it may now make sense to consider hybrid short read and long read solutions - the difference between DeepVariant at 20x and 30x is about 15,000 total errors, which is about the same number as the roughly 13,000 high quality genome in a bottle structural variants which long-read methods are especially accurate for. With noisy long reads approaching the same per-base price as short-reads, hybrid solutions may provide more research insight than higher coverage of short-read data alone.
Finally, we hope this analysis highlights how quickly the field is improving, and will continue to improve. Over time, we look forward to sequencing and analysis which is more accurate, more affordable, and with clear and favorable ways to choose the options which maximize science and discovery for the needs of a project.