Looking Through DeepVariant's Eyes
Authors:




This blog post has an accompanying Colab notebook where you can play against DeepVariant and try to beat it at its own game! First, read on to learn more about the pileup images DeepVariant uses, and then see how you do at this image classification task yourself!
DeepVariant is a deep learning-based variant caller. DeepVariant is highly accurate, the most recent version (v0.9) has SNP F1 of 0.9996 and indel F1 of 0.9981 on the PrecisionFDA Truth Challenge BAM. However, because most individuals have 4-5 million variants, at this accuracy there are still several thousand errors. There is still value in improving DeepVariant because each clinically relevant variant that it can consistently capture means that more expensive and work-intensive methods can be replaced with a single WES or WGS test. To improve DeepVariant beyond its current abilities, we need to look closely at the remaining errors.
The core concept for DeepVariant comes from how human scientists look at a putative variant in a genome browser like IGV, evaluating the evidence: How many reads support the variant? Do the reads have good base and mapping quality scores? Are there any unexpected patterns in read mapping or other variants nearby?
While DeepVariant’s concept is rooted in how humans would do the task, it is benchmarked relative to the performance of heuristic and statistical algorithms. The reason is that humans don’t scale. Looking at 4 million pileups is not anybody’s idea of a good time. While we don’t want to look at millions of pileups, it can be fun and educational to look at a few dozen of them and see if we can learn to do this classification task ourselves.
What DeepVariant sees
DeepVariant has 3 stages: make examples, call variants, and postprocess variants. The middle stage is when the deep neural network does its classification, while the first stage prepares data for the neural network, and the last stage interprets the classifications output by the neural network as variant calls.
The deep neural network is a Convolutional Neural Network (CNN) designed to classify images, so the data available at each locus is turned into a tensor that is much like an image. While images usually have three channels (red, green, and blue), DeepVariant’s examples have six channels. In this article we will show the six channels in a row, but in DeepVariant they are encoded as six layers in the third dimension, giving each tensor a shape of (100, 221, 6) corresponding to (height, width, channels). The variant in question is always in the center of each pileup image, here marked with a small line at the top.
Channels are shown in greyscale below in the following order:
-
Read base: different intensities represent A, C, G, and T.
-
Base quality: set by the sequencing machine. White is higher quality.
-
Mapping quality: set by the aligner. White is higher quality.
-
Strand of alignment: Black is forward; white is reverse.
-
Read supports variant: White means the read supports the given alternate allele, grey means it does not.
-
Base differs from ref: White means the base is different from the reference, dark grey means the base matches the reference.

For each tensor, DeepVariant’s CNN outputs three genotype likelihoods, corresponding to how many copies (0, 1, or 2) of the given alternate allele are present. Most loci are biallelic, meaning there is one putative alternate allele in addition to the reference. In biallelic loci, a classification of 0 means homozygous reference, 1 means heterozygous, and 2 means homozygous alternate (i.e. two alternate alleles are present). For example, the CNN may look at the pileup above and output (2.076e-07, 0.99999964, 1.5e-07), where the likelihood of a “1” is very high, and the “postprocess variants” stage will interpret that as “heterozygous”. We will show a multiallelic example later, but for now, let’s build up familiarity through some simpler biallelic examples first.
Here are 3 examples that we would consider canonical easy-to-classify loci, and that DeepVariant calls confidently and correctly:

The variant above is a “2”, which means both chromosomes match the variant allele, so this locus represents a homozygous alternate locus.

DeepVariant correctly classifies the variant above as a “1”, which means that one of the two alleles matches the variant allele, i.e. it is heterozygous.

This is a “0”, which means that there are no copies of the alternate allele present, and therefore this locus is homozygous reference. While it has a few supporting reads (channel 5) that caused it to be flagged as a candidate, we see that the mapping qualities (channel 2) of those reads are poor, and they have multiple mismatches (channel 6).
All examples in this blog post are from HG002 with read alignments and truth labels from the NIST Genome in a Bottle (GIAB) Consortium. See the DeepVariant WGS case study documentation for a detailed description of this dataset.
Now try these:
A.

B.

C.

D.

E.

More difficult examples
Most loci turned out to be pretty easy for us, and for DeepVariant, so we picked out all the loci that DeepVariant either got wrong or where it was less than 90% sure of its choice. That makes it a lot more interesting because we skip the 99.8% of variants that would be easy for both us and DeepVariant to get right.
Here is a selection of these “difficult” alleles. Try them yourself, then check the answers at the bottom of this post.
F.

G.

H.

I.

J.
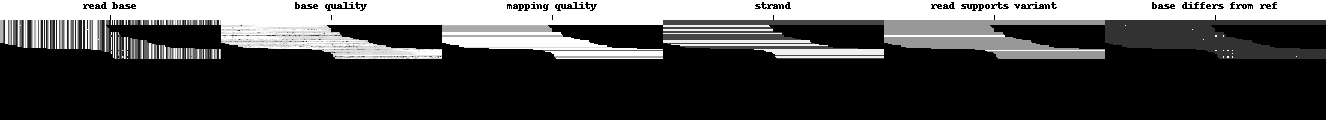
K.

L.

M.

N.
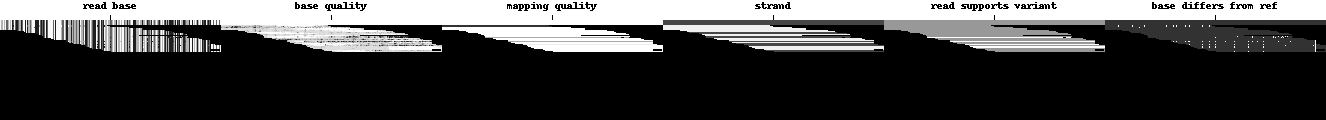
O.

Multiallelic variants
We started with biallelic variants above because they are simpler – there is only one pileup tensor for each locus, and we are only asking about a single alternate allele. For multiallelic loci, one tensor is created for each combination of either one or two alternate alleles, and the likelihoods from classifying each of those are combined.
For example, a locus with two alternate alleles yields three pileups: alt1, alt2, and alt1/alt2.
alt1: C -> CATTTT. Classification = 1

alt2: C -> CATTTTATTTT. Classification = 1

alt1/alt2: C -> CATTTT/CATTTTATTTT. Classification = 2

DeepVariant’s CNN looks at these three tensors separately and makes a call for each one, only consolidating the likelihoods into a variant call in the “postprocess variants” stage. For this example above, the classifications are alt1: 1, alt2: 1, and alt1/alt2: 2. Think of these classifications as a count of how many of the indicated allele is present, where the indicated allele corresponds to the one supported by the white reads in the fifth channel (“read supports variant”). Indeed, only the fifth channel is different between these three tensors, so it is an indication to DeepVariant of which allele to consider in multiallelic cases. This example is a case where the final variant call should be alt1/alt2, meaning one copy of each variant.
Most multiallelic cases are triallelic like the one above, but the pattern continues at higher numbers of alleles. E.g. for three alternate alleles: alt1, alt2, alt3, alt1/alt2, alt1/alt3, alt2/alt3. The following example has three alternate alleles, so we can see this pattern in action. Here the alleles and the truth classifications are shown, but remember that the CNN only sees the tensor with no additional information.
alt1: CT -> C. Classification = 1

alt2: CT -> CTT. Classification = 1

alt3: CT -> TTT. Classification = 0

alt1/alt2: CT -> C/CTT. Classification = 2

alt1/alt3: CT -> C/TTT. Classification = 1

alt2/alt3: CT -> CTT/TTT. Classification = 1

The C/CTT combination gets a 2 here, and the others are consistent in the sense that each time C or CTT show, the pileup is classified as a 1.
If you want to try some multiallelic examples, the Colab notebook has sections of easy and difficult multiallelic examples you can play.
Could we beat DeepVariant?
No, we couldn’t consistently beat DeepVariant using its own pileup images. Sometimes we would get 7/10 right when it gets 6, but the next time it’s the opposite. Overall it comes out to DeepVariant being slightly better than us, which makes sense because it has learned from millions of examples, and we haven’t.
If you have tried going up against DeepVariant in the Colab notebook you may be saying, “this is not fair, I would do much better if I could use
<my favorite genome browser>!” Well, you can! The bam file is at
gs://deepvariant/case-study-testdata/HG002_NIST_150bp_50x.bam[.bai], and the
reference is gs://deepvariant/case-study-testdata/hs37d5.fa, so feel free to
load that into your favorite genome browser and look up each locus as you go
through the game in the Colab. You can even reference other datasets, just not
the GIAB truth VCFs – no cheating! If you can beat DeepVariant using extra
information that would be generally available for other samples, then maybe we
can use these insights to train a more accurate DeepVariant model.
If a human can beat DeepVariant then knowing how can point us in a direction for improving it. Even if we can’t beat DeepVariant, then just getting used to looking at these pileup images makes it easier for us to reason about how it works and to get ideas for experimenting with the representation.
While we couldn’t beat DeepVariant ourselves, we found that trying gave us a better intuition for how DeepVariant works. Perhaps a little friendly human vs. AI competition can help us work together even better.
Did you beat DeepVariant?
Tell us about your experience going up against DeepVariant by tweeting @marianattestad.
Answers
Easy: A:1, B:0, C:1, D:2, E:0.
Difficult: shown as truth with [DeepVariant’s answer in brackets].
F:1[0], G:0[1], H:0[0], I:0[1], J:1[1], K:0[2], L:2[0], M:1[1], N:1[0], O:2[2]