Improving Variant Calling using Haplotype Information
Authors:

 Alexey Kolesnikov
Alexey Kolesnikov

In this blog, we discuss a new channel in DeepVariant which encodes haplotype information in long-read data, and was released with DeepVariant v1.1. We review how haplotypes relate to variant calling, show examples improved by the channel, and quantify the accuracy improvement with PacBio HiFi reads.
Introduction
Each individual inherits 23 pairs of chromosomes, one set from their mother and the other from their father. Genetic variation on each chromosome represents a haplotype, or group of “linked” alleles that are inherited from a single parent. Whether two parts of sequence are on the same haplotype (called cis) or opposite (trans) is important in gene regulation and interpreting the impact of variants.
In the process of sequencing, the genome is sampled in small pieces, which results in a loss of long-range haplotype information. However, long-read sequencing approaches cover large enough regions to allow local reconstruction of which variants in the genome are on the same haplotype (in phase). Because human experts often use information about parental inheritance to interpret variants, we reasoned that this information would improve accuracy of DeepVariant.
DeepVariant classifies the genotype of candidate variant positions by representing attributes of the sequence data as a tensor. The tensor is essentially a multidimensional image, consisting of channels that correspond to sequence features such as read base, base quality, and mapping quality. Each channel is centered on the candidate and has a width of 221 bp. This genomic interval provides enough local context to achieve high-accuracy with variant calling.

The width of this window was originally determined for Illumina short reads. However, long-reads contain distant variant sites outside of the window that can be used to infer haplotypes (through a process called phasing). In the context of variant calling, haplotype information can provide evidence for or against a putative variant by linking similar evidence together, as opposed to random errors. Consider the example below of heterozygous variants flanking a homopolymorphic site where haplotype information provides support for a variant.
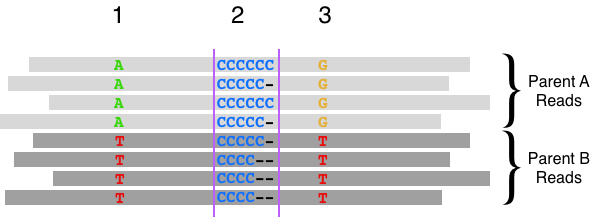
Previously, we reported that sorting reads by haplotype reduced false negatives and positives by ~30% in Wenger 2019 (and discussed in this blog post). These improvements were added to the DeepVariant 1.0 release. Notably, these improvements used an implicit representation of haplotype. We decided to investigate whether a haplotype channel, which provides a more direct representation of haplotype information, may further improve accuracy.
Approach
Integrating the Haplotype Channel
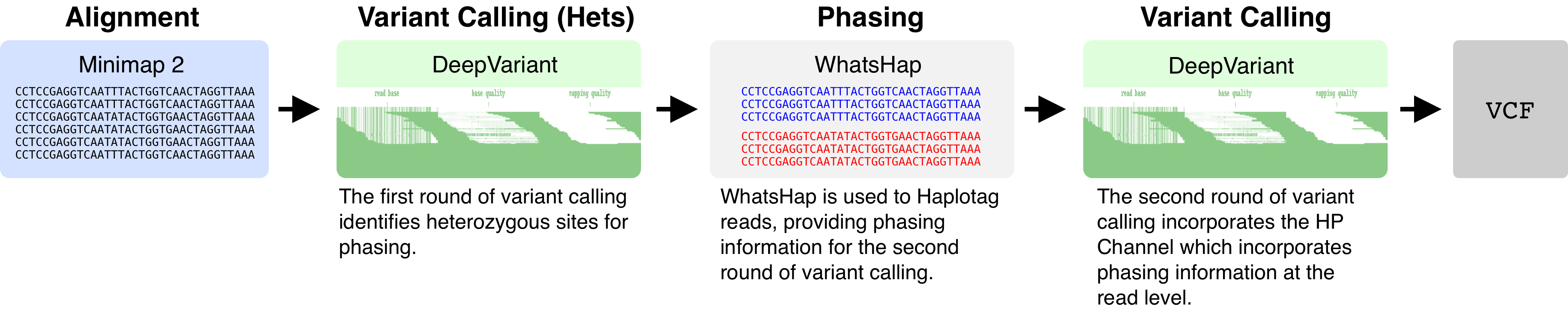
To add a haplotype channel to DeepVariant, we perform two rounds of variant calling. The first round of variant calling identifies heterozygous variants. These heterozygous variants are used by WhatsHap to perform phasing. WhatsHap will then annotate individual reads with an HP tag (a “haplotag”). The HP tag takes on values of 0, 1, or 2. HP=1 and HP=2 distinguish haplotypes at a particular locus, whereas HP=0 indicates that no haplotype could be inferred. DeepVariant fetches the HP tag, and then constructs the haplotype channel.

Training Models with the Haplotype Channel
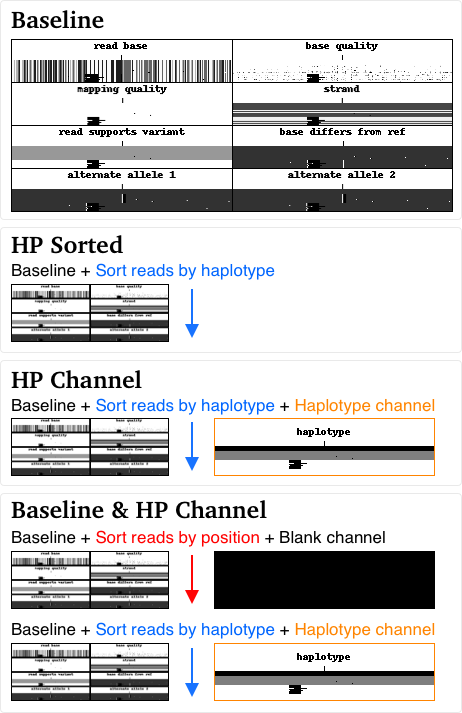
In order to test whether a haplotype channel improves accuracy using PacBio HiFi reads, we generated the following training datasets from HG001, HG002, HG004, and HG005 Genome in a Bottle (GiaB) BAMs haplotagged using WhatsHap:
- Baseline: the existing model (no haplotype information included)
- HP Sorted: Reads sorted by haplotype (similar to Wenger et al. 2019)
- HP Channel: Reads sorted by haplotype and a haplotype channel.
- Baseline & HP Channel: Our baseline training set was combined with the HP Channel training set.
The first 3 experiments are designed to give a clear test of individual hypotheses. The last experiment is designed to compare with our production training process, as our release model needs to perform well on both haplotagged and untagged data. Having a single model that operates on both types of data also means only one model is needed to perform both rounds of variant calling when integrating haplotype information.
Results
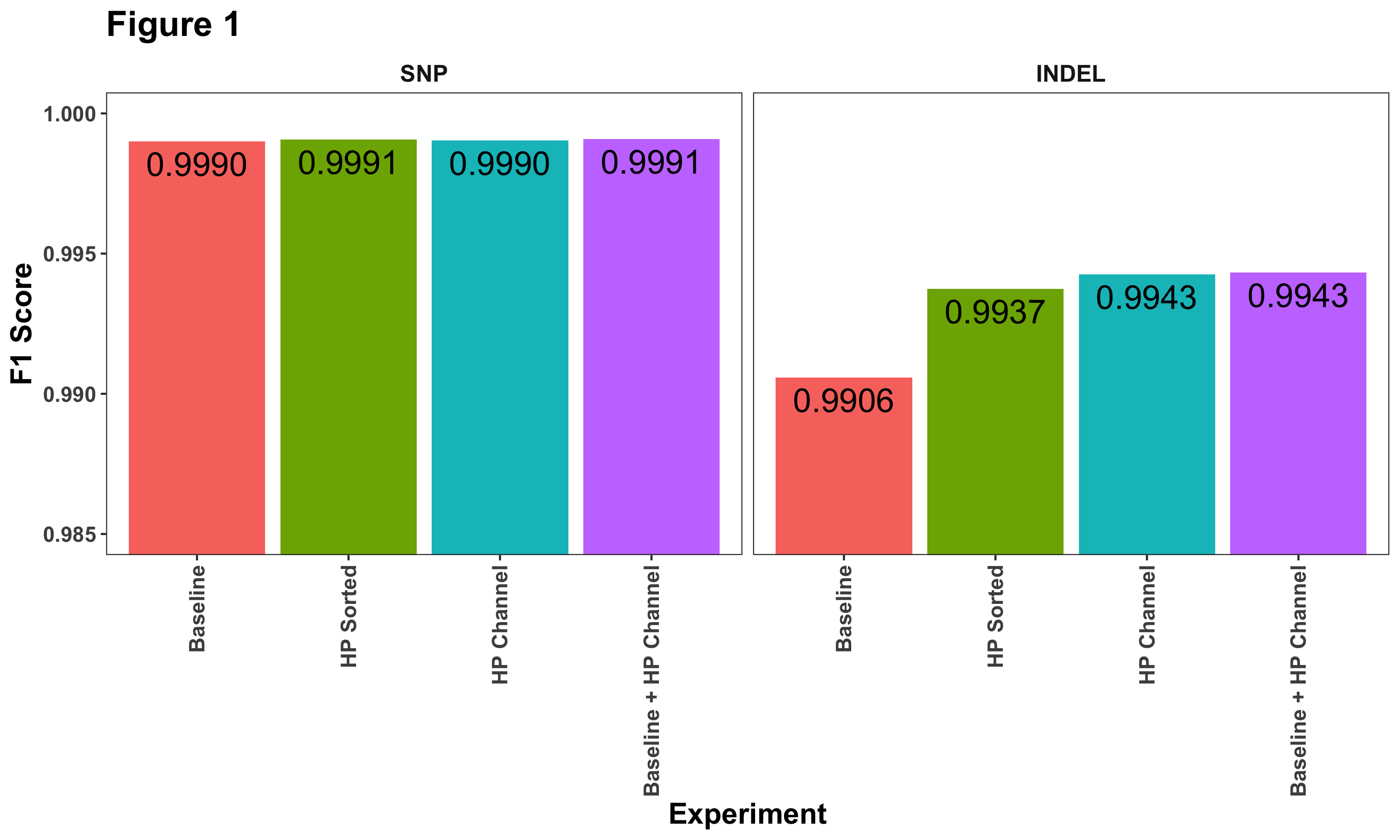
Overall, we observe the greatest improvements in INDEL calling (Figure 1). Sorting reads by haplotype drives significant improvement, but the addition of the haplotype channel further improves F1 scores.
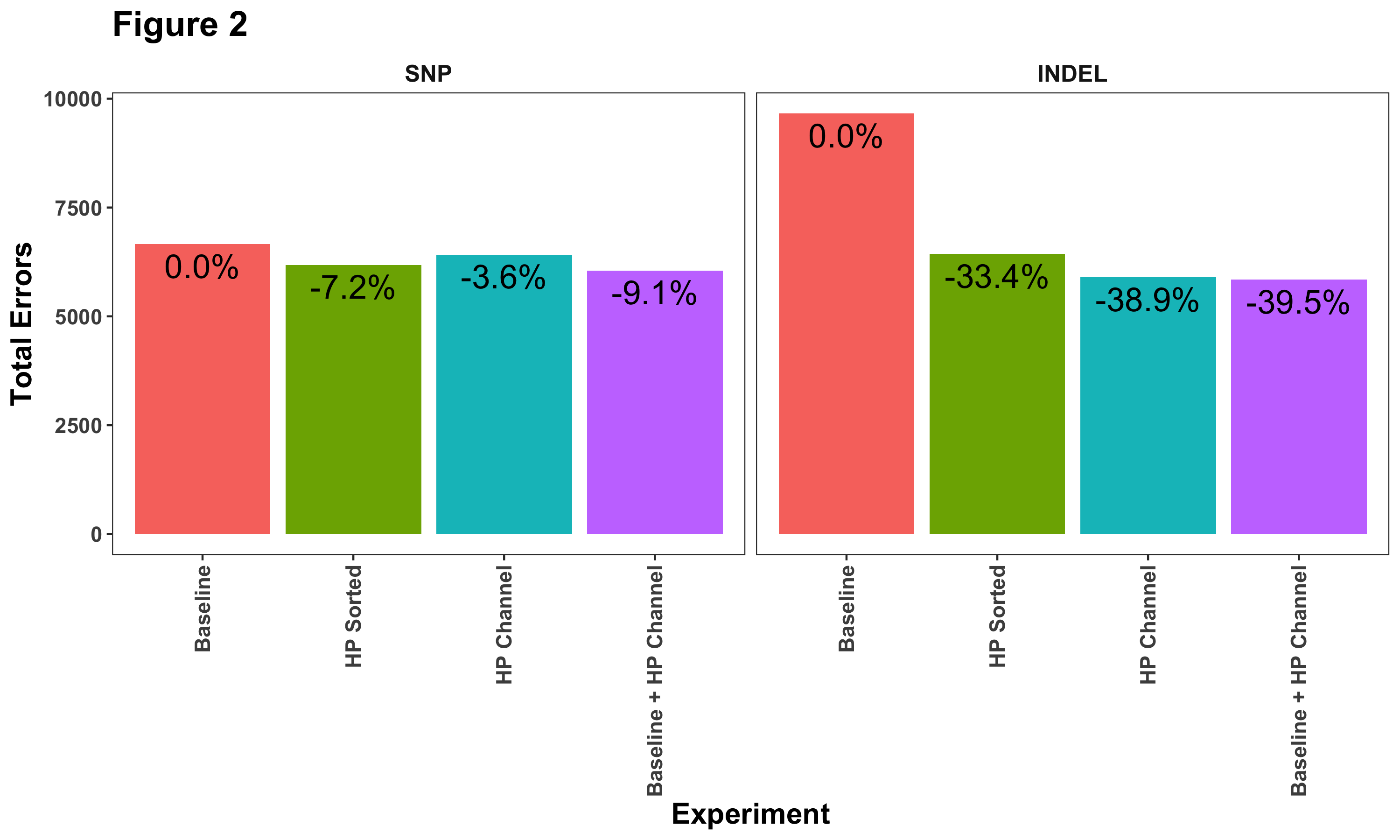
Another way to look at these results is to examine their relative improvement compared to the baseline. Figure 2 shows the total number of errors (false positives + false negatives) identified when we compare DeepVariant with the truth set. Each bar lists the percent decrease compared to our baseline model. Here we can see more clearly that haplotype information does help reduce SNP calling errors.
We reason that homopolymer INDEL errors are the main challenge in accurately calling variants in HiFi data. A variant caller has to distinguish the measurement variation in length (e.g. how many 7T or 8T runs) from the probability that one parent has 7Ts and the other 8Ts). By separating the signal by haplotype, the distributions become clearer. Labelling their boundaries with the explicit channel, and giving the variant caller clear information that the variant could be phased improves this further.
Duplicate Training Comparison
One concern we had during the addition of the HP Channel was whether results would differ considerably between training runs. To test this, we performed training and evaluation twice. The resulting evaluations did not differ more than 1.8% in terms of their total number of errors.
Runtime
We evaluated runtime using the DeepVariant PacBio case study. The addition of the haplotype channel does not have a significant effect on runtime.
| Stage | Baseline (minutes) | –use_hp_information |
|---|---|---|
| make_examples | 113m | 116m |
| call_variants | 218m | 222m |
| postprocess_variants (with gVCF) | 73m | 69m |
| total | 404m = 6.7 hours | 407m = 6.7 hours |
Conclusion
Here we have demonstrated that the addition of a haplotype channel improves the accuracy of variant calling on PacBio HiFi data. These improvements have been integrated as part of the DeepVariant 1.1 release. See the PacBio HiFi case study for details on how to call variants using haplotype information.
This work is an example of how we can improve accuracy of analysis methods by using domain intuition about a problem and representing that information in a manner that a neural network can learn. Looking forward, we will investigate additional signals including mapping percent and base quality of long reads to see whether additional new channels lead to further improvements in accuracy. We look forward to continuing to improve the accuracy of DeepVariant for our users in future releases.