Improved non-human variant calling using species-specific DeepVariant models
Authors:




Abstract
In this work, we investigate variant calling across a pedigree of mosquito (Anopheles gambiae) genomes. Using rates of Mendelian violation, we assess pipelines developed to call variation in humans when applied to mosquito samples. We demonstrate the ability to rapidly retrain DeepVariant without the need for a gold standard set by using sites that are consistent versus inconsistent with Mendelian inheritance. We show that this substantially improves calling accuracy by tuning DeepVariant for the new genome context. Finally, we generate a model for accurate variant calling on low-coverage mosquito genomes and a corresponding variant callset.
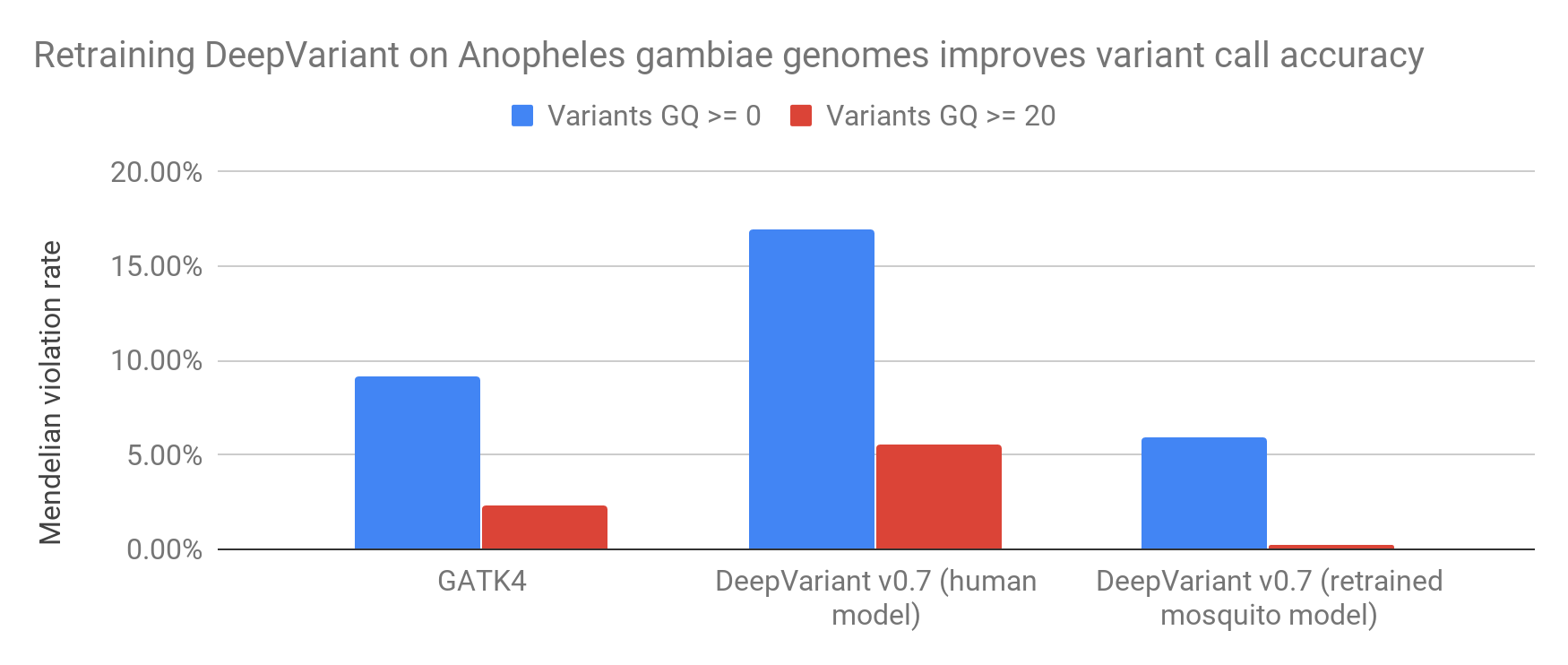
Figure 1: Summary: retraining DeepVariant improves accuracy of variant calling in mosquito genomes.
Introduction
Many of the first population-scale genomics projects were performed on human samples. Tool development has been strongly influenced by this. GATK, for example, suggested a set of known human indels when it included indel realignment in its best practices, and still includes the use of the human dbSNP resource in the BQSR step of its best practices. In addition, human variation is shaped by properties and a history not shared across many organisms. The short generation time and large number of progeny in many species suggest a different ability to generate, tolerate, and select on variation when compared to humans. This can result in genomic signatures that might require a different set of priors to analyze in an ideal manner.
The MalariaGEN project uses genomics to understand how population genetics in humans, mosquitoes, and Plasmodium relate to the transmission, prevention, and treatment of malaria. This project has generated medium-coverage whole genome sequencing (WGS) data on mosquito populations from the Ag1000G dataset. This also contains large pedigrees (currently 80 samples from 4 crosses), which allows us to assess and ultimately improve tools for genomic analysis applied in mosquitoes.
Applying standard variant calling methods on mosquito genomes without modification
First, we sought to understand the performance of existing methods on the MalariaGEN data by calculating Mendelian errors on trio data. Specifically, we started with this trio:
Mother: AD0231-C; Father: AD0232-C; Child: AD0234-C.
Our first set of experiments was to run the basic WGS settings of
DeepVariant v0.7 and
GATK4 HaplotypeCaller
directly. This created single-sample callsets for each of the three individuals.
For DeepVariant, we merged these callsets into a cohort VCF containing the three
individuals using GLnexus with the
--config DeepVariant flag, which creates a unified representation of variants
but does not perform any filtering or joint calling of genotypes. For GATK4
HaplotypeCaller, we followed GATK’s “best practices” workflow for germline short
variant discovery, by running CombineGVCFs to consolidate gVCFs and
GenotypeGVCFs to perform joint genotyping. Due to the lack of a mosquito dbSNP, GATK4 BQSR cannot be used here. All statistics are calculated on
the non-mask regions of the genome, though there is little difference when mask
regions are included.
In contrast to humans, where the Genome in a Bottle Consortium (GiaB) has developed a set of gold standard variant calls in multiple individuals, we do not have ground truth data for the mosquito variant calls. We can estimate the accuracy of the callsets as a whole using Mendelian inheritance violations as a measure of the error rate.
Evaluating the performance of current pipelines on mosquito genomes.
The initial analysis comparing the GATK and DeepVariant results used the open-source RealTimeGenomics RTG Tools to identify Mendelian violations. These are stratified by the genotype quality of the call (minGQ), which is a caller-produced estimation that the genotype is correct.
Table 1: Variant counts and Mendelian violations for GATK4 and DeepVariant v0.7 WGS model. Human Baseline is from HG002, hs37d5.
| GATK4 | |||
|---|---|---|---|
| Variants analyzed | Mendelian Errors | Mendelian Errors rate | |
| minGQ=0 | 7,110,329 | 653,955 | 9.20% |
| minGQ=20 | 5,414,888 | 123,406 | 2.28% |
| Human Baseline minGQ=0 |
7,210,317 | 620,770 | 8.61% |
| Human Baseline minGQ=20 |
6,526,680 | 289,493 | 4.44% |
| DeepVariant v0.7 | |||
|---|---|---|---|
| Variants analyzed | Mendelian Errors | Mendelian Errors rate | |
| minGQ=0 | 4,634,748 | 784,051 | 16.92% |
| minGQ=20 | 1,691,825 | 93,484 | 5.53% |
| Human Baseline minGQ=0 |
6,254,576 | 180,191 | 2.88% |
| Human Baseline minGQ=20 |
5,720,100 | 96,741 | 1.69% |
There are multiple points of interest in Table 1.
- Both methods exhibit higher rates of Mendelian violation in total calls (GQ>=0) in the mosquito data compared to the human baseline, suggesting an accuracy gap.
- GATK4 reports many more total variants than DeepVariant.
- At GQ>=20, GATK4 reports over three times as many variants as DeepVariant. Note that estimates of genotype quality are not necessarily comparable across callers as they may be computed in substantially different ways. An alternative scaling would hold the total number of reported variants constant.
- The Mendelian violation rate for GATK4 is lower than that of DeepVariant, which is unusual as DeepVariant has a much lower Mendelian violation rate for human data.
Based on these results, we hypothesized DeepVariant uses properties in the genome structure of humans which are powerful for variant calling, but are different in mosquitoes.
Investigating why DeepVariant accuracy differs in mosquitoes
We investigated the actual calls when a Mendelian violation occurs, and found that they skew dramatically toward records in which the child mosquito has a homozygous reference (HOM_REF) call. We examined the homozygous reference calls and their supporting evidence in the child mosquito since they were enriched for Mendelian violations.
There are two ways that an individual can be called as homozygous reference at a position in DeepVariant. 1) There is little or no evidence of any variation at the site, so no example is created and evaluated by the convolutional neural network (CNN). 2) The site has enough variation that an example is created and evaluated by the CNN, and the most likely of the three states (HOM_REF, HETEROZYGOUS, HOM_ALT) predicted is HOM_REF.
Of the 94,554 Mendelian violations where the child is HOM_REF, only 17,475 (18%) of those have the HOM_REF call based just upon reference and non-reference read counts, the remaining 82% had the HOM_REF call produced by the CNN. This seemed suspicious, so we investigated the allele depth fractions for each of HOM_REF, HETEROZYGOUS, and HOM_ALT calls in all three individuals (Figure 2). While the HETEROZYGOUS and HOM_ALT variant types have distributions of alternate allele fractions that are consistent with the genotype calls, the HOM_REF allele distributions are much different than expected, with a large fraction of the HOM_REF calls containing nearly all non-reference reads at the location. (Note that the absence of HOM_REF calls with only reference reads is expected; those sites do not generate candidate variants to be evaluated by the CNN). The presence of HOM_REF calls with mostly non-reference reads remained even when filtering to calls with genotype quality >= 20.
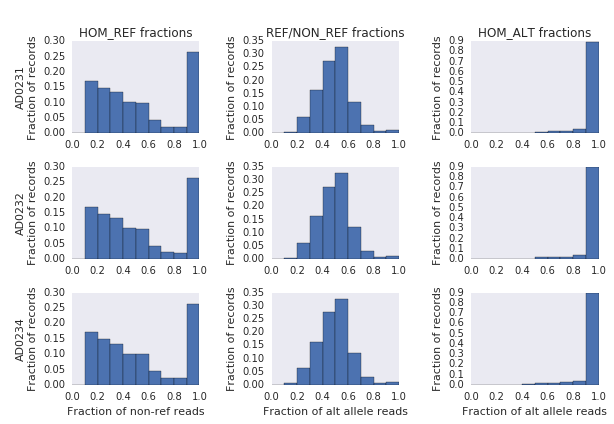
Figure 2: Allele depth fractions for each individual at the three common variant types.
To understand this phenomenon better, we examined individual sites that were confidently predicted as HOM_REF but composed of many non-reference reads. A representative example is shown in Figure 3.
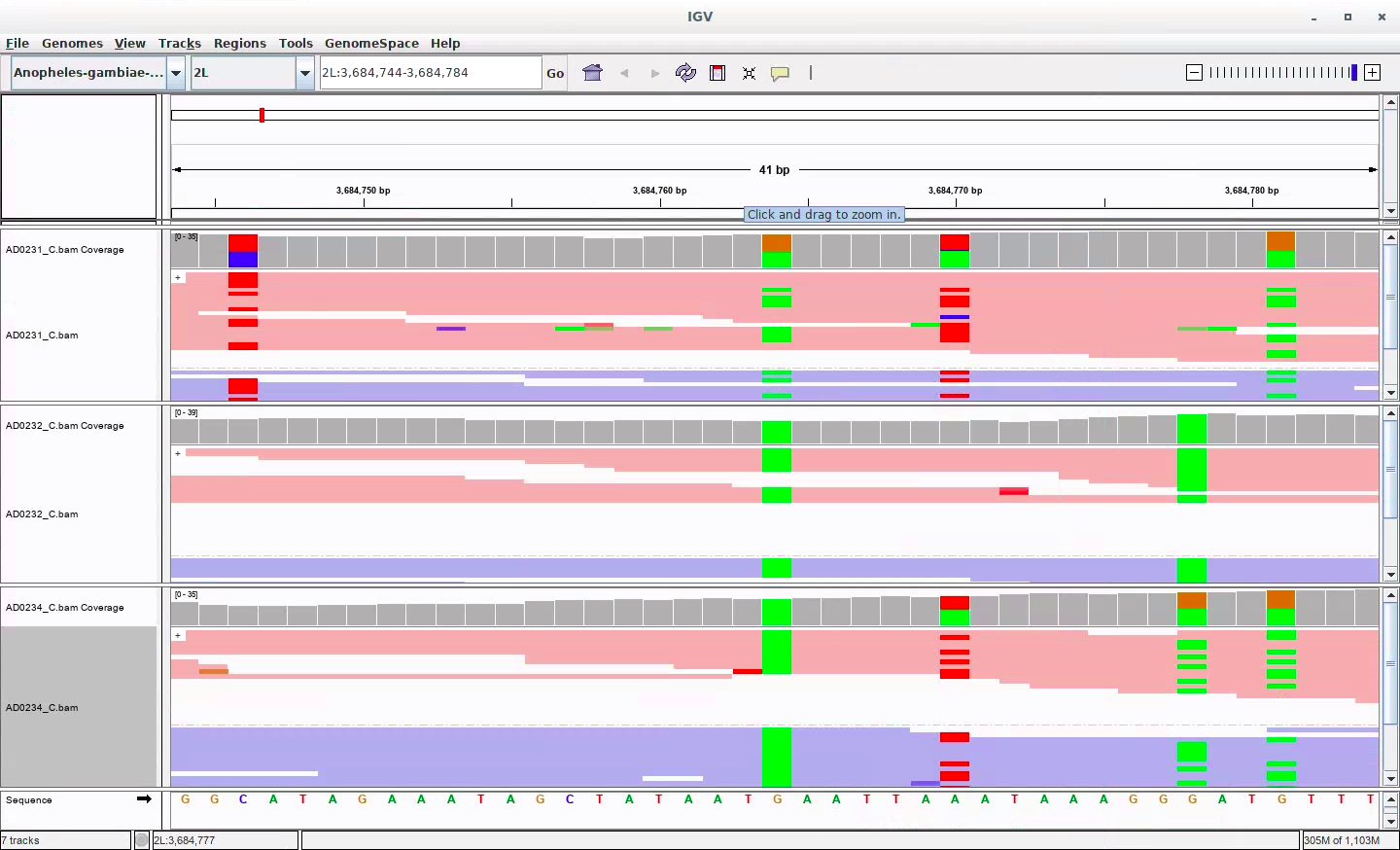
Figure 3: A representative Mendelian violation. Reads from AD0231-C (mother), AD0232-C (father), and AD0234-C (child) are shown from top to bottom. The corresponding calls are HOM_REF, HOM_ALT, HOM_ALT, respectively.
The mother is called as HOM_REF despite having 14 of the 30 reads covering the region support the alternate allele, suggesting misclassification of a truly heterozygous site. The segregation of variants along the reads and subsequent transmission to the child corroborate this site as a real variant. From this, it seems clear that the presence of nearby variants is a signal that a variant is suspicious in human variant calling in a way that is true to a different degree in mosquitoes.
A hypothesis for why this behavior occurs is that in humans the number of true variants seen in a single window is much lower than the number seen here, and regions where multiple putative variants occur are enriched for false positive calls. Because of the lower variant density in humans, clustered variants of this nature may be more likely to be caused by mismapping from similar genomic regions, while in mosquito genomes the higher variant density may mean a signature like this likely reflects true variants. This implies that retraining on mosquito variants could improve our model’s robustness to these data.
Designing the “curriculum” to re-train DeepVariant for the mosquito genome
To train a DeepVariant mosquito-specific model, we generated a “silver standard” callset for any child of the AD0231-C and AD0232-C mosquitoes using calls from GATK4 with additional filters. Confident HOM_REF calls for each mosquito were defined as regions with coverage of at least 15 reads, a minimum reference read count of 13, minimum reference allele fraction of 0.86. Confident HOM_ALT calls for each parent mosquito were defined as calls with coverage of at least 12 reads, a minimum alternate allele read count of 10, minimum alternate allele fraction of 0.83. The child “silver standard” callset is composed of variants for which both parents have confident calls, restricted to the non-masked regions of the mosquito genome, and contains 972,344 heterozygous variants and 560,307 homozygous alternate variants.
We trained the model using these positions as truth and the read data from 5 other progeny of AD0231-C and AD0232-C for training: AD0242-C, AD0243-C, AD0244-C, AD0245-C, AD0246-C, and then tuned the model using the read data from AD0250-C. To train a DeepVariant mosquito-specific model, we initialized the training from the v0.7 WGS model and selected the checkpoint for which the tuning set loss was smallest.
Separately, we also started from DeepVariant calls and applied training in a similar way to achieve an improved model. The ability to start from the calls of multiple methods raises an interesting promise: the ability to train DeepVariant with the information perspective of any of the diverse set of community analysis solutions. In the event an investigation reveals that a method has unique insight in a subset of the overall variant calling problem, we (or you) can produce examples to train DeepVariant to capture this insight.
To summarize from a machine learning/training perspective, we are optimizing for two properties: correctness of the label, and representativeness of the examples. To generate examples, we look for positions that we can confidently determine the transmitted allele from each parent, but do not look at the evidence in the child. This allows us to represent sites that may be difficult in the child, but which have a known label. Furthermore, we generate examples of REF, HET, and HOM ALT in approximately the same ratio that DeepVariant would see them in a real sample.
Importantly, the techniques to generate this silver standard dataset should generally apply to any case where a pedigree of individuals exists, regardless of whether it is a non-human species or human samples sequenced with a novel technology.
The DeepVariant mosquito-specific model shows significant improvement on callset quality
The final callset was generated as described for the initial v0.7 WGS model. Initial results from evaluating the DeepVariant mosquito-specific model on AD0234-C are shown in Table 2.
Table 2: Variant counts and Mendelian violations for GATK4, DeepVariant v0.7 WGS, and a DeepVariant mosquito-specific model. Note: The GATK4 and DeepVariant v0.7 WGS results are replicated from Table 1.
| GATK4 | |||
|---|---|---|---|
| Variants analyzed | Mendelian Errors | Mendelian Errors rate | |
| minGQ=0 | 7,110,329 | 653,955 | 9.20% |
| minGQ=20 | 5,414,888 | 123,406 | 2.28% |
| DeepVariant v0.7 | |||
|---|---|---|---|
| Variants analyzed | Mendelian Errors | Mendelian Errors rate | |
| minGQ=0 | 4,634,748 | 784,051 | 16.92% |
| minGQ=20 | 1,691,825 | 93,484 | 5.53% |
| DeepVariant mosquito-specific model | |||
|---|---|---|---|
| Variants analyzed | Mendelian Errors | Mendelian Errors rate | |
| minGQ=0 | 7,206,679 | 427,175 | 5.93% |
| minGQ=10 (match # variants to GATK minGQ=20) | 5,246,248 | 53,495 | 1.02% |
| minGQ=20 | 3,705,653 | 8,181 | 0.22% |
As Table 2 demonstrates, the Mendelian violation rate of the DeepVariant mosquito-specific model is lower than GATK at a comparable number of variants. To further illustrate what the difference is, we ranked the variant calls from GATK4 and from the re-trained DeepVariant model by the confidence that each caller assigned the call based on the genotype quality. This allows us a direct comparison between the accuracy at a variant-by-variant level. As Figure 4 indicates, DeepVariant is significantly more accurate overall, across all calls. DeepVariant is also able to provide a higher reliability in its most confident calls. Variants with the highest quality scores from DeepVariant have a very low Mendelian violation rate (reaching less than 0.1%), while a batch of GATK4’s most confident calls never achieve less than a ~1% Mendelian violation rate.
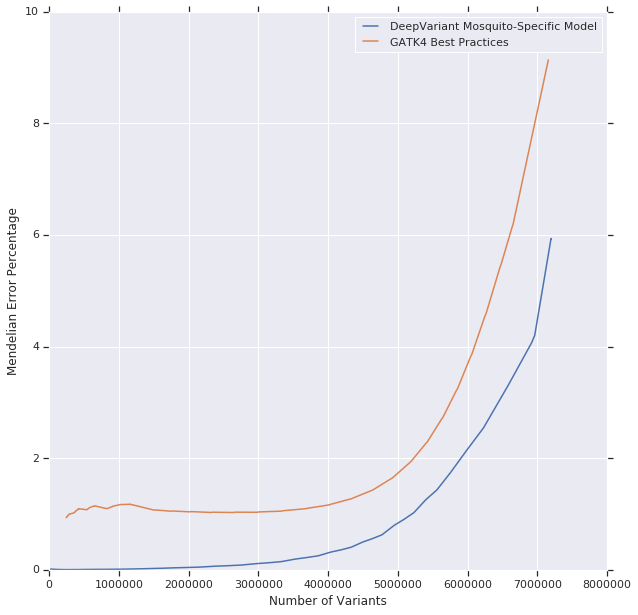
Figure 4: Number of variants and Mendelian violations in trio callsets from GATK4 and the DeepVariant mosquito-specific model, per varying levels of minGQ, based on calls in the child. A smaller number of variants corresponds to a higher minGQ threshold. So the Mendelian violation rate at 1,000,000 x-axis position is the rate seen in the top 1,000,000 variants ranked by the confidence of GATK4 or DeepVariant.
Future work
The main products of this investigation are a demonstrated ability to retrain DeepVariant for improved performance in new genomes or genomic contexts. This has generated callset of the mosquito pedigree for investigation by the community.
This initial foray into mosquito variant calling has identified multiple areas for further investigation. First, additional evaluations of the variant calls of other progeny of AD0231-C and AD0232-C will address how generalizable these results are to those samples. Second, creating “silver standard” calls for the other mosquito parents and evaluating the models on their progeny will further address the generalization capabilities of the models to the mosquito genome more broadly. Third, a deeper investigation into the causes of the confident HOM_REF calls for sites with mostly non-reference reads is warranted, which may involve exploration of alternative modeling strategies to explicitly capture prediction uncertainty.
Callsets
The final merged callsets generated by the DeepVariant mosquito-specific model
and GATK4 are publicly available in
Google Cloud Storage
under gs://brain-genomics-public/DeepVariant-blog/MalariaGEN bucket.
Instructions on accessing this dataset can be found in
Cloud Storage documentation.